


EQUIPE 1 – Philippe Charron – Génomique et physiopathologie des maladies cardiovasculaires : des maladies monogéniques aux maladies multifactorielles
- Thèmes de recherche
- Membres de l'équipe 1
- Choix de publications
- Collaborations internationales
- Partenaires financiers

Ph. Charron, responsable de l’équipe 1
CONTEXTE CLINIQUE ET SCIENTIFIQUE DES PROJETS DE RECHERCHE DE L’ÉQUIPE
Les cardiomyopathies et canalopathies, les deux sous-groupes principaux des maladies cardiaques héréditaires, sont les causes principales de mort subite cardiaque et d’insuffisance cardiaque chez les jeunes patients (<40 ans), en particulier chez les sportifs. Malgré l’amélioration de la prise en charge de ces maladies, grâce aux médicaments pharmacologiques, aux dispositifs implantables et à la transplantation cardiaque, de nouvelles connaissances sur les causes génétiques sous-jacentes, les voies métaboliques et la physiopathologie sont nécessaires pour identifier de nouvelles cibles ou stratégies thérapeutiques et pour mieux prévenir les complications dévastatrices de ces maladies.
Des avancées récentes dans la connaissance des causes de ces maladies (en particulier les variants dans des gènes sarcomériques et des canaux ioniques) ont par ailleurs conduit à une nouvelle compréhension de l’interaction complexe entre architecture génétique (variants rares, mutations, et fréquents) et interactions avec des facteurs environnementaux (comme le sport, la myocardite, les drogues) ou le sexe des patients. Ces nouvelles connaissances et ce nouveau paradigme ont des conséquences importantes pour la compréhension globale de la physiologie des protéines sarcomères et des canaux ioniques comme pour la physiopathologie de maladies complexes telles que l’insuffisance cardiaque et l’arythmie.
Depuis plus de 20 ans, notre groupe s’intéresse au décryptage des mécanismes génétiques et cellulaires sous-jacents au développement des cardiomyopathies et des canalopathies. Nous avons récemment identifié de nouveaux variants génétiques, rares ou fréquents, impliquées dans ces maladies grâce à des stratégies génomiques d’analyse d’association (GWAS) ou de séquençage. Nous étudions des voies de signalisation sous-jacentes et initions de nouvelles approches thérapeutiques à partir de ces avancées scientifiques. Simultanément, nous développons, dans la pratique clinique, des approches translationnelles comprenant des tests génétiques et un reséquençage à haut débit, afin d’améliorer la prise en charge médicale des patients et de leurs familles grâce à une médecine personnalisée.
L’équipe 1 développe ses recherches autour de 4 axes :
- Transférer les nouvelles connaissances sur la génétique et la physiopathologie en pratique clinique, notamment par le biais de tests génétiques, de reséquençage à haut débit, pour développer une médecine personnalisée
- Identifier les acteurs-clés (variants génétiques rares et fréquents, voies de signalisation) dans les cardiomyopathies et les canalopathies par des approches multi-omiques
- Comprendre la fonction, les interactions et la physiopathologie des principaux acteurs de ces maladies à travers des cardiomyocytes humains dérivés de cellules souches pluripotentes induites (iPS) et des modèles murins
- Progresser vers de nouvelles thérapies (pharmacologiques, interventionnelles, génétiques) dans les formes monogéniques ou complexes d’insuffisance cardiaque et d’arythmies.
L’équipe 1 est membre de l’École Doctorale 394 : Physiologie, Physiopathologie et Thérapeutique.
THÈME 1 – GÉNOMIQUE DE L’INSUFFISANCE CARDIAQUE CAUSEE PAR LA CARDIOMYOPATHIE DILATÉE
Éric Villard, Sophie Garnier, Richard Isnard, Philippe Charron
Antécédents. La cardiomyopathie dilatée est une maladie complexe à la fois familiale et sporadique. Démêler la composante génétique de cette maladie pour améliorer les soins aux patients et trouver de nouvelles cibles thérapeutiques est l’une des tâches principales de notre groupe. Dans les formes familiales, la stratégie d’analyse de liaison / gène candidat positionnel a été développée (Sylvius N, 2001), complétée maintenant par le séquençage des exomes et du génome de nouvelle génération. Pour les formes sporadiques, la stratégie du gène candidat était assez infructueuse et, rapidement, nous nous sommes tournés vers des études d’association à haut débit sur des milliers de cas et de témoins. Des GWAS successifs sur des échantillons d’ADN groupés (Villard E, 2011) ou en utilisant des SNP exomiques enrichis (Esslinger U, 2017) ont révélé 8 loci génomiques candidats (HSPB7, TTN, SLC39A8, MLIP, FLNC, BAG3, ALPK3 et FHOD3), deux étant également impliqués dans l’insuffisance cardiaque ischémique (Garnier S, 2015). Un troisième GWAS sur 2 700 cas sporadiques de DCM et 4 400 témoins, combiné à une analyse d’imputation, est actuellement en cours pour identifier de nouveaux loci de susceptibilité.
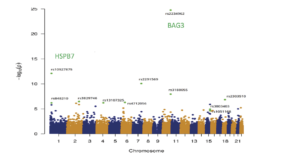
Manhattan Plot – Exome Wide Association Study. 95.499 variants were investigated by logistic regression analysis. Associations are summarized in the Manhattan plot which displays (green dots), the eleven SNVs significantly associated with DMC. From Esslinger U and al. 2017
Notre groupe est également très impliqué dans l’impact pathologique des gènes découverts. Sur la base de nos résultats de GWAS/EWAS dans le DCM, nous émettons l’hypothèse que le contrôle de la qualité de la protéine, dans le contexte de contraintes mécaniques répétées, est une voie majeure pour la santé des cardiomyocytes. Ces hypothèses, et les applications thérapeutiques potentielles, sont remises en question en utilisant des modèles murins knock-in de cardiomyocytes dérivés de DCM et iPs avec du tissu cardiaque engenieré (EHT) découpé à partir de cellules de patients mutées. Nous avons également initié un projet collaboratif avec l’industrie pharmaceutique dont le but est d’identifier les voies moléculaires impliquées dans la cardiomyopathie hypertrophique et de réaliser un criblage moléculaire fondé sur des cardiomyocytes dérivés de l’IP. Nous bénéficions de notre ancrage fort au sein du centre national de référence pour les maladies cardiaques héréditaires et d’un accès unique aux patients et à leurs génotypes, de la structure interne dédiée aux iPs-cardiomyocytes et de nos compétences en édition génomique.
THÈME 2 – Analyse génomique de la réponse anormale de repolarisation cardiaque aux médicaments pharmacologiques
Joe-Elie Salem, Christian Funck-Brentano
De nombreux médicaments utilisés à des fins non cardiovasculaires ou cardiovasculaires, comme le sotalol, ont pour effet indésirable d’allonger la durée de la repolarisation cardiaque en inhibant le canal potassique IKr (KCNH2), ce qui peut déclencher des arythmies cardiaques potentiellement mortelles. Les modifications des paramètres ECG de la repolarisation (tels que QTc, Tpeak-Tend, Tamp, déformations de l’onde T) varient considérablement d’un sujet à l’autre, suggérant l’existence de facteurs génétiques prédisposants. Dans l’étude GENEREPOL, les modifications de la repolarisation induites par une dose orale de sotalol ont été analysées chez 990 sujets en bonne santé. QTc et TpTe ont augmenté et TAmp a diminué. Un sous-échantillon aléatoire de 489 individus a été soumis à une analyse d’association pangénomique où 8 306 856 polymorphismes nucléotidiques uniques (SNP) imputés ont été testés pour leur association avec les variations de QTc, TpTe et TAmp (D), ainsi que leurs composants principaux dérivés, et l’apparition de déformations de l’onde T. Aucun des SNP étudiés n’a atteint le seuil statistique (Salem JE et al, 2017). Cependant, l’étude a montré qu’une analyse des composants principaux basée sur les variations de QTcF, TAmp et TpTe pourrait être un moyen intégratif de différencier davantage les patients ayant une inhibition extrême d’IKr. Nous avons breveté cet algorithme de diagnostic en 2016 comme méthode de détermination de la susceptibilité à l’induction d’une torsade de pointes (PCT / EP2017 / 058714, BRV 116 – WO – 2016-096).
Notre projet actuel vise à comprendre les déterminants de la repolarisation ventriculaire dépendant du sexe. La repolarisation cardiaque est influencée par des interactions complexes entre les hormones stéroïdes sexuelles et les gonadotrophines (Salem JE et al, 2016), selon le sexe. Nous avons récemment montré que le rapport progestérone / estradiol chez la femme, la testostérone chez l’homme et la FSH dans les deux sexes sont des déterminants majeurs de la repolarisation ventriculaire (Abehsira G et al, 2016). Nous prévoyons d’étudier plus avant dans la cohorte GENEREPOL l’influence des hormones sexuelles sur l’allongement de l’intervalle QT induit par les médicaments, associée à l’utilisation du séquençage de l’exome entier à la recherche de variants rares et de basse fréquence. Nous rechercherons également une interaction entre les hormones et l’expression de gènes à l’aide de cardiomyocytes dérivés de cellules iPS de patients atteints du syndrome du QT long congénital ou de QT longs médicamenteux. Nous prévoyons de tester si une administration hormonale exogène appropriée pourrait augmenter la réserve de repolarisation en situation à risque. Nous avons déjà breveté une méthode de traitement des « torsades de pointes » par administration hormonale exogène (PCT / EP2017 / 059097, BRV 119 – WO – 2016-120).
THEME 3 – From deciphering the genetic determinant of ventricular arrhythmia to therapeutic applications
Nathalie Neyroud, Fabrice Extramiana, Antoine Leenhardt, Pascale Guicheney
Our group has been interested for many years in deciphering the genetic background of cardiac channelopathies (Blancard M 2018, Clatot J 2012). The SCN5A gene encodes the human cardiac Na+ channel α-subunit, Nav1.5, responsible for initiation and propagation of the action potential. Loss-of-function mutations in SCN5A have been linked to life-threatening cardiac arrhythmias (such as the Brugada syndrome or sick sinus syndrome (Ziyadeh-Isleem A 2014). A murin model of Scn5a haplo-insufficiency has been developed, partially recapitulating the Brugada syndrome phenotype. Our project sought to overexpress the cardiac Na+ channel in Scn5a+/- deficient mice in an attempt to restore their Na+ current and their ECG parameters. As a consequence of the large size of the SCN5A gene, we have developed a dual AAV vector strategy to produce two AAV9 populations allowing, in vivo, the translation of the full hSCN5A gene fused to the gfp gene as a reporter. Eight weeks after systemic AAV-injection in mice, our results show a robust transduction of cardiac cells, a normalization of the PR interval on injected-mice ECGs, and a significant increase of the Na+ current in transduced myocytes (Doisne et al., manuscript in preparation). These results will give impulse to gene therapy strategies of malignant arrhythmias observed in SCN5A loss-of-function-related channelopathies.
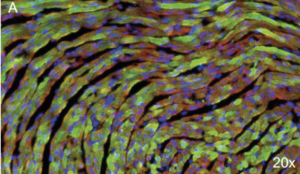
Eight-micrometer slices of heart tissue from a mouse injected with the AAV carrying hSCN5A. AAV injection led to the expression of Nav1.5 tagged with the GFP in almost 50% of heart cells. A. Nuclei are stained in blue, a-actinin in red and the GFP in green (20x).
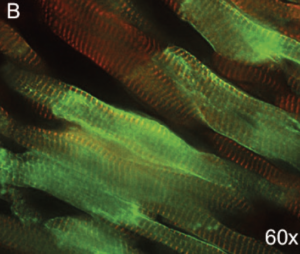
B. Magnification of transduced cardiomyocytes (60x).
Idiopathic Ventricular Fibrillation (IVF) is a rare cause of sudden cardiac arrest. The exact incidence of IVF is unknown but is declining with the advance of diagnostic testing and the discovery of primary arrhythmia syndromes (Visser M, 2016 Circ. Arrhythm. Electrophysiol). For more than 15 years, our group has worked to investigate patients presenting with ventricular tachycardia and to collect their familial information and DNA. We have recently selected a group of 60 cases diagnosed with IVF originating from Purkinje fibers. All were resuscitated cardiac arrest patients in whom known cardiac, respiratory, metabolic, and toxicological etiologies had been excluded. Our project seeks to identify new genes and variants involved in IVF by a whole-exome sequencing approach.
THEME 4 – Identify new physiological pathways in Arrhythmogenic right ventricular cardiomyopathy using innovative human cellular models
Estelle Gandjbakhch, Eric Villard
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a rare inherited cardiomyopathy characterized by fibrofatty replacement of myocytes leading to ventricular arrhythmia, sudden death and heart failure mainly caused by desmomosal genes mutations (Fressard V, 2010). Ongoing work by our team has shown the essential role of cadherin mediated adhesion in the disease (Vite A,2013 & Vite A., submitted). One major challenge in translational research of ARVC is to generate a pertinent cardiac cellular model expressing mature desmosomes and recapitulating cyclic mechanical load. In this project, we will generate an IPS-derived engineered heart tissue (hIPS-EHT) which will be used as an innovative in vitro model to unravel physiopathology of ARVC and explain the mutation specific phenotypes. This 3D cardiac model reconstitutes a mature contractile cardiac tissue reproducing mechanical stress, as observed in vivo. We aim to structurally compare the effect of PKP2 and DSG2 mutations on cardiomyocyte and junction structures using 3D immunofluorescent imaging as well as their consequences on desmosome function and cardiomyocytes electrophysiological properties. We also aim to study the expression profiles associated with PKP2 and DSG2 mutations by whole-transcriptome analysis using high throughput RNA sequencing.
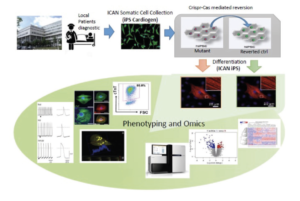
Production of human cardiomyocytes derived from patient iPS cells with mutation of interest
THEME 5 – Translational impact of genetic testing on precision medicine
Pascale Richard, Véronique Fressart, Estelle Gandjbakhch, Philippe Charron
Apart from the use of genetic results in the cascade screening of families with hereditary diseases (Charron P, 2010 & Cirino AL, 2017), our team has reported several publications about the role of some genes or mutations in the severity of cardiac diseases, such as the role of multiple mutations on the prognosis of a cardiomyopathy (Richard P, 2002, Fressard V, 2010) or the role of some particular genes such as PRKAG2 & GLA in HCM (Thevenon J, 2016, Séné T, 2016) or DSG2 in ARVC (Hermida A, in press). Identification of these genetic factors may help to identify high risk profile patients that would benefit from close cardiac monitoring and early treatments such as implantable cardioverter defibrillator or heart failure therapy. An illustrative example of direct personalized therapeutic impact is about defibrillator implantation decision based on genetics. Our team actively participated to international joint efforts to establish the independent prognostic role of LMNA mutation, associated with high risk of sudden death, and to clarify the identification of patients that would benefit most from implantable cardiac defibrillator implantation (van Rijsingen IA, 2012; Kumar S, 2016; Thuillot M, 2019).
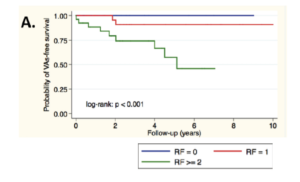
First validation of a prognostic score in heart failure due to LMNA. Four predicitve factors (non sustained ventricular tachycardia. LV Ejection Fraction <45%, male sex and non-missense mutations) for malignant ventricular arrythmia (VA) were studied in a monocentric cohort. ICD is recommended in the presence of 2 risk factors. From Thuillot M. & al., 2019.
Refine the genetic spectrum of cardiomyopathies with high throughput sequencing. Large cohorts of patients with Left ventricle non-compaction (LVNC), HCM and DCM have been collected through the national network of the Referral center for cardiac hereditary diseases. Resequencing strategy with various panels of genes, including a panel of >100 genes involved in various cardiac hereditary diseases are under way in order to refine the prevalence of genes, the overlap between cardiomyopathy subtypes and then progress towards optimal diagnostic strategies for routine care of patients and families.

| Name | Position | ORCID |
|---|
- Hermida, A, Ader, F, Millat, G, Jedraszak, G, Vogel, L, Garçon, L et al.. RBM20 Gene in Patients With Cardiomyopathy: Phenotypic Expression for Loss-of-Function Versus Hotspot Variants. Circ Heart Fail. 2025;18 (3):e012492. doi: 10.1161/CIRCHEARTFAILURE.124.012492. PubMed PMID:39823286 .
- Jurgens, SJ, Rämö, JT, Kramarenko, DR, Wijdeveld, LFJM, Haas, J, Chaffin, MD et al.. Genome-wide association study reveals mechanisms underlying dilated cardiomyopathy and myocardial resilience. Nat Genet. 2024;56 (12):2636-2645. doi: 10.1038/s41588-024-01975-5. PubMed PMID:39572784 PubMed Central PMC11631763.
- Zheng, SL, Henry, A, Cannie, D, Lee, M, Miller, D, McGurk, KA et al.. Genome-wide association analysis provides insights into the molecular etiology of dilated cardiomyopathy. Nat Genet. 2024;56 (12):2646-2658. doi: 10.1038/s41588-024-01952-y. PubMed PMID:39572783 PubMed Central PMC11631752.
- Ader, F, Jedraszak, G, Janin, A, Billon, C, Buisson, NR, Bloch, A et al.. Prevalence and phenotypes associated with ALPK3 null variants in a large French multicentric cohort: Confirming its involvement in hypertrophic cardiomyopathy. Clin Genet. 2024;105 (6):676-682. doi: 10.1111/cge.14505. PubMed PMID:38356193 .
- Ferrand, MC, Giordano, G, Mougenot, N, Laporte, PL, Vignier, N, Leclerc, A et al.. Intracardiac electrophysiology to characterize susceptibility to ventricular arrhythmias in murine models. Front Physiol. 2024;15 :1326663. doi: 10.3389/fphys.2024.1326663. PubMed PMID:38322613 PubMed Central PMC10846502.
- Perret, C, Proust, C, Esslinger, U, Ader, F, Haas, J, Pruny, JF et al.. DNA-pools targeted-sequencing as a robust cost-effective method to detect rare variants: Application to dilated cardiomyopathy genetic diagnosis. Clin Genet. 2024;105 (2):185-189. doi: 10.1111/cge.14427. PubMed PMID:37904629 .
- Kato, K, Isbell, HM, Fressart, V, Denjoy, I, Debbiche, A, Itoh, H et al.. Novel CALM3 Variant Causing Calmodulinopathy With Variable Expressivity in a 4-Generation Family. Circ Arrhythm Electrophysiol. 2022;15 (3):e010572. doi: 10.1161/CIRCEP.121.010572. PubMed PMID:35225649 .
- Gizon, M, Duboscq-Bidot, L, El Kassar, L, Bobin, P, Ader, F, Giraud-Triboult, K et al.. Generation of a heterozygous SCN5A knockout human induced pluripotent stem cell line by CRISPR/Cas9 edition. Stem Cell Res. 2022;60 :102680. doi: 10.1016/j.scr.2022.102680. PubMed PMID:35093717 .
- Blancard, M, Touat-Hamici, Z, Aguilar-Sanchez, Y, Yin, L, Vaksmann, G, Roux-Buisson, N et al.. A Type 2 Ryanodine Receptor Variant in the Helical Domain 2 Associated with an Impairment of the Adrenergic Response. J Pers Med. 2021;11 (6):. doi: 10.3390/jpm11060579. PubMed PMID:34202968 PubMed Central PMC8235491.
- Doisne, N, Grauso, M, Mougenot, N, Clergue, M, Souil, C, Coulombe, A et al.. In vivo Dominant-Negative Effect of an SCN5A Brugada Syndrome Variant. Front Physiol. 2021;12 :661413. doi: 10.3389/fphys.2021.661413. PubMed PMID:34122134 PubMed Central PMC8195286.
- Garnier, S, Harakalova, M, Weiss, S, Mokry, M, Regitz-Zagrosek, V, Hengstenberg, C et al.. Genome-wide association analysis in dilated cardiomyopathy reveals two new players in systolic heart failure on chromosomes 3p25.1 and 22q11.23. Eur Heart J. 2021;42 (20):2000-2011. doi: 10.1093/eurheartj/ehab030. PubMed PMID:33677556 PubMed Central PMC8139853.
- Touat-Hamici, Z, Blancard, M, Ma, R, Lin, L, Iddir, Y, Denjoy, I et al.. A SPRY1 domain cardiac ryanodine receptor variant associated with short-coupled torsade de pointes. Sci Rep. 2021;11 (1):5243. doi: 10.1038/s41598-021-84373-9. PubMed PMID:33664309 PubMed Central PMC7970841.
- Vite, A, Gandjbakhch, E, Hery, T, Fressart, V, Gary, F, Simon, F et al.. Desmoglein-2 mutations in propeptide cleavage-site causes arrhythmogenic right ventricular cardiomyopathy/dysplasia by impairing extracellular 1-dependent desmosomal interactions upon cellular stress. Europace. 2020;22 (2):320-329. doi: 10.1093/europace/euz329. PubMed PMID:31845994 .
- Richard, P, Ader, F, Roux, M, Donal, E, Eicher, JC, Aoutil, N et al.. Targeted panel sequencing in adult patients with left ventricular non-compaction reveals a large genetic heterogeneity. Clin Genet. 2019;95 (3):356-367. doi: 10.1111/cge.13484. PubMed PMID:30471092 .

