


EQUIPE 3 – Elise Balse et Sophie Nadaud – Plasticité cellulaire et moléculaire dans les maladies cardiovasculaires
- Thèmes de recherche
- Membres de l'équipe 3
- Choix de publications
- Collaborations
- Prix
- Partenaires financiers

Elise Balse & Sophie Nadaud, Team 3 Heads
Objectifs de notre recherche
Les maladies cardiovasculaires sont souvent le résultat de modifications profondes des propriétés fonctionnelles et structurelles du cœur et des vaisseaux sanguins, causées par divers facteurs individuels et environnementaux. Différents mécanismes moléculaires et cellulaires communs au cœur et aux vaisseaux sanguins sous-tendent ce processus de remodelage. Notre équipe vise à comprendre les acteurs de la plasticité moléculaire et cellulaire qui caractérise le remodelage cardiovasculaire dans le contexte de différentes maladies cardiovasculaires : fibrillation auriculaire (FA), insuffisance cardiaque (IC) et hypertension artérielle systémique (HTA) et pulmonaire (HP). Ces maladies sont les principales causes de mortalité et de morbidité dans le monde et partagent des caractéristiques épidémiologiques, notamment dans les maladies métaboliques telles que le diabète de type 2, la MAFLD ou l’obésité.
Tous nos projets sont soutenus par l’environnement d’excellence scientifique et technologique de l’IHU ICAN (Institut Hospitalo-Universitaire de Cardiométabolisme et Nutrition). En particulier, nos études sont menées sur aux plateformes techniques et scientifiques de l’IHU ICAN : ICAN-BioCell iPS pour la production de cellules dérivées d’iPS, ICAN– Omics pour les études métabolomiques et lipidomique, ICAN-I/O pour la modélisation mathématique, les statistiques ou l’intelligence artificielle.
Nos projets portent sur
– la plasticité de la composition cellulaire des tissus cardiovasculaires. Nous étudions notamment la capacité des cellules souches et progénitrices à être recrutées, à se différencier en diverses lignées cellulaires mésenchymateuses et à contribuer au remodelage auriculaire et vasculaire.
– la plasticité des complexes protéiques macromoléculaires régulant la fonction cardiaque et leur rôle dans le dysfonctionnement de la pompe et les arythmies. Nous nous concentrons sur la régulation du trafic et de l’adressage des canaux ioniques dans les cardiomyocytes.
– le rôle des maladies métaboliques dans la régulation du remodelage vasculaire et cardiaque .
– le rôle de facteurs natriurétiques dans le contexte de l’insuffisance cardiaque chez les sujets diabétiques.
– le rôle des cellules immunitaires et inflammatoires lors du développement de la cardiomyopathie atriale.
– l’utilisation de l’Intelligence artificielle pour mieux comprendre et prédire la physiopathologie cardiovasculaire et identifier de nouveaux groupes de patients ou facteurs de risque.
Précédentes découvertes
Au cours des dernières années, notre équipe a identifié divers mécanismes impliqués dans le remodelage cardiaque ou vasculaire. Nous avons montré des altérations de la dynamique des canaux ioniques dans les cardiomyocytes dans le contexte du remodelage auriculaire et de la fibrillation auriculaire. Nous avons identifié des cellules progénitrices épicardiques qui participent au remodelage auriculaire conduisant à la fibrillation auriculaire. Nous avons également identifié des cellules progénitrices vasculaires qui sont activées lors de l’hypertension pulmonaire conduisant au remodelage vasculaire
THÉMATIQUES
THEME 1 – Plasticité des cardiomyocytes : adaptations structurelles et fonctionnelles pathologiques – Elise BALSE
Membres du groupe
Elise BALSE, PhD, MCUHC, Maître de Conférence à Sorbonne Université
Gilles DILANIAN, Ingénieur d’Etude
Dounia FAHRI, Doctorante
Athina KOUMANTAROU, Doctorante
Contexte scientifique et contributions passées
L’expression fonctionnelle des canaux ioniques dans le sarcolemme des cardiomyocytes détermine la forme et la durée du potentiel d’action, contrôlant ainsi la période réfractaire effective du myocarde. L’expression fonctionnelle correcte des canaux ioniques peut être perturbée à plusieurs niveaux, notamment au niveau transcriptionnel, traductionnel et post-traductionnel. Au cours des dernières années, nous avons contribué à une meilleure compréhension de la dynamique de l’expression de surface des canaux ioniques dans les myocytes cardiaques, en particulier le canal de repolarisation spécifique des oreillettes KV1.5 dans le contexte du remodelage auriculaire et de la fibrillation auriculaire (Figure 1) (Balse et al. PNAS 2009, Balse et al., Physiol Rev 2012 ; Boycott et al., PNAS 2013 ; Melgari et al., JMCC 2020). Nous avons également étudié la fonction des membres de la famille des protéines MAGUK dans la régulation des canaux potassiques cardiaques (El-Haou et al., 2009 : Abi-Char et al., Amer. J Physiol. 2008) et du canal sodique cardiaque (Eichel et al., Circ Res 2016 ; Beuriot et al. Heart Rhythm 2020) (Figure 2).
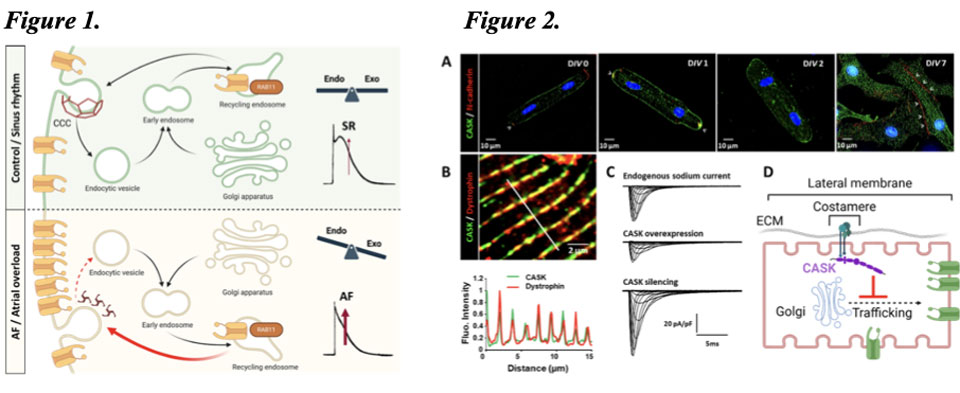
Figure 1. Labilité de l’expression de surface du canal KV1.5. (En haut) Les processus de trafic intracellulaire antérograde et rétrograde sont en équilibre dans des conditions physiologiques. (En bas) Dans des situations pathologiques, telles qu’une surcharge hémodynamique auriculaire chronique ou une fibrillation auriculaire, les processus d’exocytose prédominent, entraînant une accumulation des canaux KV1.5 à la surface des cardiomyocytes et un raccourcissement du potentiel d’action auriculaire. Figure 2. La protéine MAGUK CASK, un partenaire non conventionnel du canal sodique cardiaque NaV1.5. A) La CASK est une protéine spécifiquement exclue du disque intercalaire , comme le montre son exclusion du marquage avec la protéine N-cadhérine. B) CASK colocalise avec la dystrophine au niveau du costamère des cardiomyocytes. C) Contrairement à d’autres partenaires identifiés, CASK régule négativement le courant sodique. D) Schéma récapitulatif montrant que CASK régule l’expression fonctionnelle du canal en agissant sur son trafic antérograde.
Parmi les protéines MAGUK, nous accordons une attention particulière à la protéine CASK, qui semble réguler, outre le canal sodique, divers composants du connexome tels que les protéines des jonctions communicantes et des desmosomes dans des conditions physiologiques et dans le contexte de la cardiomyopathie arythmogène du ventricule droit (CAVD) (Blandin et al., bioRxiv 2024) (Figure 3).
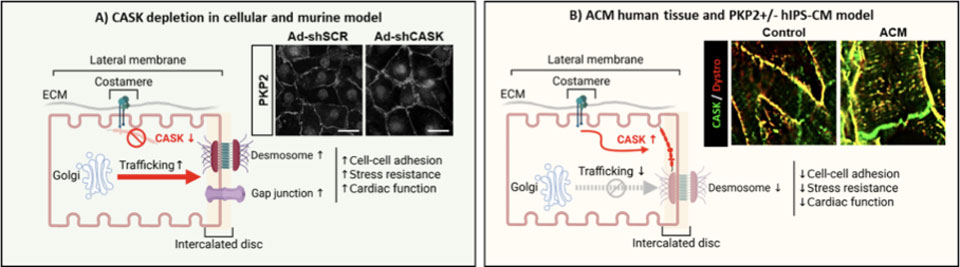
Figure 3. CASK, le contrôleur du trafic des protéines du connexome. A) La déplétion de CASK favorise le transport des protéines du connexome (connexine 43 et plakophiline-2) vers le disque intercalaire dans différents modèles cellulaires (cardiomyocytes néonataux, cardiomyocytes dérivés de cellules souches pluripotentes induites, hIPS-CM). Dans un modèle cellulaire porteur d’une mutation desmosomale responsable d’une CAVD (PKP2+/- hIPS-CM), la déplétion en CASK améliore la cohésion cellulaire et la résistance au stress. Chez le rat, la déplétion en CASK par vecteur AAV favorise la fonction cardiaque et la contractilité. B) Dans les biopsies de patients atteints de CAVD, CASK est relocalisée au niveau du disque intercalaire et son expression est augmentée.
Projets en cours
Nos travaux nous ont amenés à nous intéresser plus particulièrement au lien entre les propriétés électriques et l’organisation tridimensionnelle des cardiomyocytes, en particulier les microdomaines membranaires, dans des contextes physiologiques et physiopathologiques. Nos travaux actuels s’articulent autour de deux axes de recherche :
– Déterminer le rôle de la signalisation membranaire latérale, et plus particulièrement de la protéine CASK, dans l’organisation et le maintien du disque intercalaire au cours du développement cardiaque postnatal et dans le contexte de la CAVD.
– Identifier les altérations développementales de la structure des cardiomyocytes induites par un régime alimentaire maternel obésogène conduisant à une insuffisance cardiaque chez la progéniture à l’âge adulte.
Outils
Nous utilisons des modèles murins (rats, souris) pour diverses études fonctionnelles (échocardiographie Doppler, relation pression-volume, ECG) afin d’étudier les effets d’une stratégie d’invalidation du gène CASK à l’aide de vecteurs AAV et la fonction cardiaque des descendants nés de mères obèses ayant suivi un régime riche en graisses. Nous utilisons des modèles cellulaires in vitro allant des cultures de cardiomyocytes primaires aux cellules dérivées d’hIPSC (cardiomyocytes, progéniteurs épicardiques), saines ou porteuses de mutations causales de la CAVD. Dans ces modèles in vitro, nous utilisons des vecteurs adénoviraux pour surexprimer ou invalider l’expression des protéines d’intérêt. Notre expertise comprend l’électrophysiologie classique (patch-clamp), l’imagerie moléculaire et tissulaire à haute résolution. Nos recherches s’appuient également sur des approches omiques (RNAseq, protéomique, métabolomique, lipidomique) et des collaborations avec des cliniciens de l’IHU ICAN.
Importance
Nos recherches visent à mieux comprendre les mécanismes de trafic et d’adressage des protéines dans les microdomaines membranaires des myocytes cardiaques afin de mettre en évidence les réserves de plasticité cardiaque dans les maladies héréditaires telles que la CVDA, ou les maladies acquises telles que l’insuffisance cardiaque.
Collaborations
Collaborations Internationales:
Imperial College London, J. GORELIK
Collaborations Nationales:
Institut Pasteur Paris, T. WEI
I2MC, Toulouse, C. GALES
THEME 2 – Plasticité cellulaire et moléculaire lors du remodelage vasculaire dans l’hypertension artérielle, l’hypertension artérielle pulmonaire et le développement d’une maladie veino-occlusive pulmonaire – Sophie NADAUD & Florent SOUBRIER
Membres du groupe
Sophie NADAUD, PhD, Chercheuse CRHC INSERM
Florent SOUBRIER, MD, PhD, PUPH émérite
Fabrice ATASSI, Ingénieur d’Etude
Thin Hinan NABET, Doctorante
Erika LAURO, Doctorante
Lylia HAKEM, étudiante en Master2
Ritej MAHJOUB, étudiante en Master2
Contexte scientifique et contributions passées
Notre groupe s’intéresse aux acteurs cellulaires et moléculaires impliqués dans la régulation du remodelage vasculaire. Les altérations structurelles des petits vaisseaux (principalement les artères) jouent un rôle majeur dans le développement de l’hypertension pulmonaire et systémique.
Hypertension pulmonaire :
1/ Nous avons étudié l’origine des nouvelles cellules musculaires lisses produites lors de l’hypertension artérielle pulmonaire. Il s’agit d’une maladie rare et dévastatrice, sans option thérapeutique curative, caractérisée par un remodelage occlusif de la vasculature pulmonaire distale qui conduit à une insuffisance cardiaque droite. Les vaisseaux non musculaires se muscularisent et l’épaisseur de la média artérielle augmente, avec en outre la formation d’une néointima. Les progéniteurs pulmonaires résidents participent au remodelage vasculaire associé à l’hypertension pulmonaire en générant de nouvelles cellules musculaires lisses. Nous avons identifié des progéniteurs résidents CD34+/PW1+/PDGFRα+ impliqués dans la néomuscularisation précoce observée lors d’une hypoxie chronique (CH : un modèle d’hypertension pulmonaire modérée) (Dierick, Circ Res 2016, Bordenave, ATVB 2020). Nous avons récemment démontré que la prolifération de ces cellules progénitrices est sous le contrôle de la voie PDGFRα (Solinc, JAHA 2022). Figures 4, 5, 6.
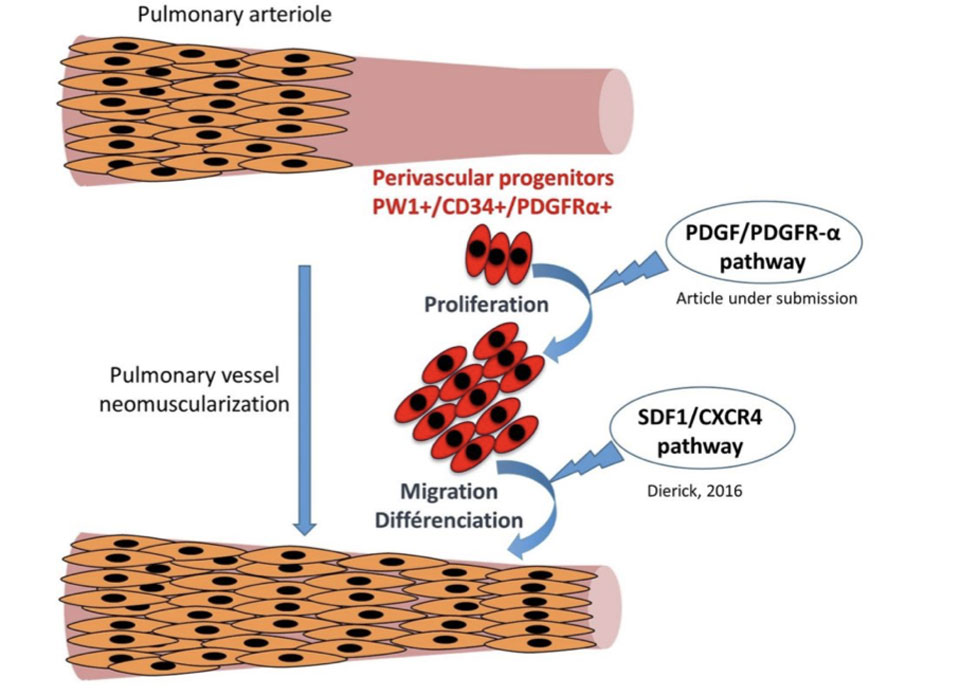
Figure 4. Les artérioles pulmonaires sont principalement non muscularisées et la néomuscularisation est une caractéristique de l’hypertension pulmonaire. Nos données montrent que les cellules progénitrices vasculaires pulmonaires sont recrutées par l’activation de la signalisation PDGFRα et se différencient en nouvelles cellules musculaires lisses à la suite de la signalisation CXCR4

Figure 5. Les cellules progénitrices en culture se différencient en cellules musculaires lisses identifiées par l’expression de SM-MHC (rouge) et de α-SMA (vert).
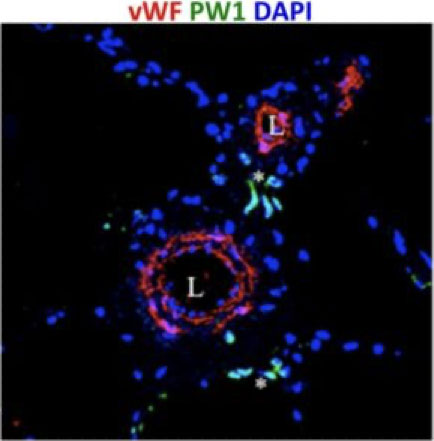
Figure 6. Les cellules progénitrices PW1+ (en vert) sont regroupées autour des vaisseaux (en rouge) dans un poumon humain contrôle.
2/ Notre groupe, en étroite collaboration avec le laboratoire de génétique clinique de l’hôpital universitaire (hôpital de la Pitié-Salpêtrière), a identifié des gènes impliqués dans la prédisposition génétique à l’hypertension artérielle pulmonaire (HAP) et à la maladie veino-occlusive pulmonaire (MVOP) : BMP10 et KDR comme nouveaux gènes responsables de l’HTAP héréditaire (Eyries, Eur Resp J 2019) et EIF2AK4 (GCN2) comme gène responsable de la PVOD (Eyries, Nat Genet 2014), Figure 7. Le lien entre la perte complète de GCN2, une sérine-thréonine kinase, et la PVOD n’est pas élucidé et fait l’objet de recherches intensives. Nous avons généré des rats porteurs de délétions de EIF2AK4 obtenues par ciblage génétique CrispR-CAS9 afin de déchiffrer, in vivo, les modes d’initiation de la maladie. Nous avons démontré que dans des conditions basales et en cas de privation d’asparagine et de glutamine induite par l’administration d’asparaginase, les rats Gcn2-/- présentent des signatures moléculaires et cellulaires dans les poumons qui pourraient indiquer un rôle de Gcn2 dans l’homéostasie immunitaire et fournir des indices supplémentaires sur les mécanismes de développement de la PVOD héréditaire (Bignard, Am J Physiol Lung Cell Mol Physiol, 2023).
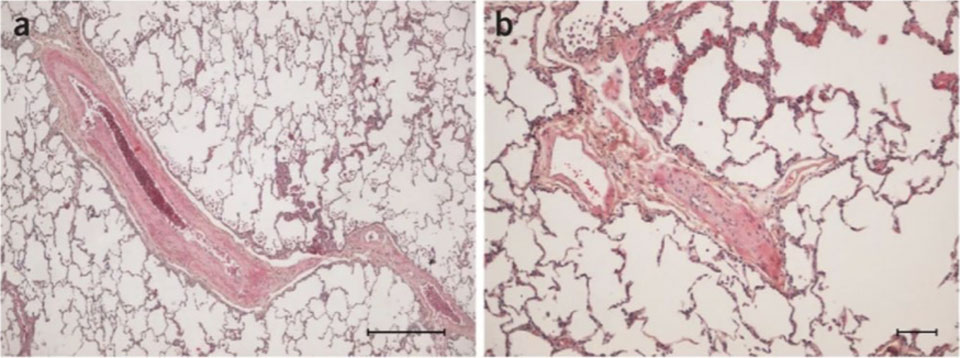
Figure 7. Pathologie de la PVOD héréditaire. (a) Veine septale et (b) petite veine présentant une fibrose intimale et un épaississement de la paroi vasculaire. D’après Eyries and al. PMID: 24292273
Projets en cours
Vaisseaux pulmonaires :
1/ En collaboration avec l’ASNR (Autorité de Sûreté Nucléaire Française), nous étudions les effets de faibles doses de rayonnements ionisants sur le remodelage et la fonction des vaisseaux pulmonaires. Ces faibles doses peuvent affecter les personnes vivant dans des zones contaminées ou les travailleurs du nucléaire. Des études épidémiologiques ont montré une augmentation des maladies cardiovasculaires dans les populations exposées à de faibles doses de rayonnements, cette augmentation devenant plus prononcée avec l’âge. Nous étudions également comment la réponse vasculaire varie en fonction de l’âge sur des souris jeunes et âgées afin de reproduire l’exposition humaine.
2/ Le récepteur du virus SARS-CoV-2, l’ACE2 (enzyme de conversion de l’angiotensine 2), est une protéine exprimée dans les cellules vasculaires. Nous avons lancé un projet utilisant des souris transgéniques exprimant l’ACE2 humaine (hACE2) afin d’étudier les effets de l’infection par le SARS-CoV-2 sur les vaisseaux pulmonaires et de déterminer les voies qui sont activées.
Vaisseaux systémiques :
3/ Nous étudions actuellement l’effet de l’obésité et du syndrome métabolique sur le remodelage des petites artères et le développement de l’hypertension systémique. Notre objectif est de déchiffrer les mécanismes impliqués dans la production de nouvelles cellules musculaires lisses lors de maladies métaboliques. Notre hypothèse est que ce remodelage est lié à l’activation des cellules progénitrices périvasculaires. Nous étudions ces cellules progénitrices et les voies de signalisation qui conduisent à leur recrutement pour former de nouvelles cellules musculaires lisses dans les maladies métaboliques. La prévention de ce remodelage pourrait contribuer à réduire l’augmentation de la pression artérielle observée dans ces conditions. Nous étudierons des modèles murins, mais également des échantillons humains provenant de patients hypertendus diabétiques et non diabétiques.
Outils
Modèles murins : nous développons des modèles de traçage de lignage cellulaire afin de suivre le devenir des cellules progénitrices lors du remodelage vasculaire associé à l’hypertension. Nous utilisons des modèles murins transgéniques pour induire ou supprimer l’activité des voies candidates in vivo après activation du tamoxifène afin d’évaluer leur rôle dans la fonction de ces progéniteurs et dans le remodelage vasculaire induit par un régime riche en graisses. Nous effectuons des mesures hémodynamiques de la pression artérielle et pulmonaire ainsi que des analyses échographiques & Doppler de la fonction vasculaire et cardiaque. Nous utilisons un régime riche en graisses pour induire l’obésité, le syndrome métabolique et l’augmentation de la pression artérielle.
Nous analysons les altérations vasculaires à l’aide d’une imagerie fluorescente des coupes tissulaires. Nous étudions le devenir des cellules à l’aide de la cytométrie en flux et de l’imagerie tissulaire. Nous utilisons des installations pour effectuer des analyses transcriptomiques de cellules individuelles et l’isolement et l’analyse de cellules par cytométrie en flux. Nous étudierons les échantillons de patients à l’aide de l’imagerie par fluorescence ainsi que de la transcriptomique spatiale. Les variations de l’expression des protéines sont également étudiées à l’aide de biochimie classique (western-blot et ELISA) classique et de la RT-qPCR.
Collaborations
Collaborations Internationales :
Victor Chang Cardiac Research Institute, Darlinghurst, Australia, R.P. HARVEY, P. SCHOFIELD, D. CHRIST
Mc Gill University, Canada, F. DIERICK
Collaborations Nationales :
MITOVASC, Angers, D. HENRION, E. VESSIERES, L. LOUFFRANI
U999, Le Kremlin Bicêtre, C. GUIGNABERT, M. HUMBERT
Institut de Biologie Paris Seine IBPS, Paris, T. JAFFREDO
Autorité de Sûreté Nucléaire et de Radioprotection ASNR, Fontenay aux Roses, V. MONCEAU
Institut Desbrest d’Epidémiologie et de Santé Publique, Département de Virologie APHP, Paris, A.-G. MARCELIN, S. MAROT
Vascular Surgery department, APHP, Paris, T. COUTURE
UMS28, Paris, S. MOROSAN
UMRS 1166, Paris, F. FOUFELLE, O. BOURRON
THEME 3 – Cardiomyopathie auriculaire et physiopathologie de la fibrillation auriculaire – Nadine SUFFEE
Membres du groupe
Nadine SUFFEE, PhD, Chercheuse CRCN INSERM
Stéphane HATEM, PUPH
Eva TRENQUIER, Assistante Ingénieur
Raoul MANUEL, PhD, Post-Doctorant
Fouad FSEIL, étudiant en Master2
Ibrahim ABOU-SNEIM, étudiant en Master2
Contexte scientifique et contributions passées
Au cours de la dernière décennie, des études cliniques ont établi que l’accumulation de tissu adipeux à la surface de l’oreillette est un facteur de risque majeur de fibrillation auriculaire (FA), en particulier lors de troubles métaboliques tels que l’obésité (Hatem et al JACC 2016). Une analyse histomorphométrique a révélé deux groupes de composants auriculaires, l’un étant l’accumulation de tissu adipeux épicardique (TAE) et l’autre étant l’infiltration fibrotique qui remplace le TAE (Figure 8A). Nous avons identifié des adipokines (Activine A) sécrétées par le TAE auriculaire humain qui favorisent la fibrose dans l’épicarde atrial (Venteclef et al EHJ 2015). Nous avons ensuite décrit comment le TAE devient fibrotique dans l’oreillette, un processus de remodelage associé à une FA persistante et médié par l’infiltration de cellules immunitaires (Haemers et al EHJ 2017). Nos recherches sur l’origine cellulaire des adipocytes et des myofibroblastes qui composent respectivement le tissu adipeux épicardique et la fibrose ont révélé une source commune provenant des cellules progénitrices nichées dans la couche épicardique (EPDC, cellules dérivées des progéniteurs épicardiques) (Figure 8B) (Suffee et al PNAS 2017). Nous avons montré un phénotype hétérogène des EPDC (Figure 8C) qui mène à leur différenciation en adipocytes ou en myofibroblastes en réponse à des facteurs locaux (Suffee et al Circ Res 2020). La faible concentration de peptide natriurétique auriculaire (ANP) sécrétée par les myocytes auriculaires est un puissant facteur adipogénique pour les EPDC exprimant le récepteur NPRA via la voie de signalisation GMPc/PKG (Suffee et al PNAS 2017). De plus, l’axe AGTR1/Smad2 exprimé dans les EPDC induit la voie de fibrogénèse en réponse à l’angiotensine-2 (Suffee et al Circ Res 2020). L’accumulation de TAE est médiée par un facteur préadipocytaire, Pref-1, exprimé sur les EPDC. Le mécanisme de Pref-1 est transitoire et dépend de facteurs locaux. Les EPDC-Pref1+ sont réactivées par un stress métabolique tel que les acides gras libres qui engagent ce précurseur dans la voie de l’adipogenèse (Azevedo et al, en cours).
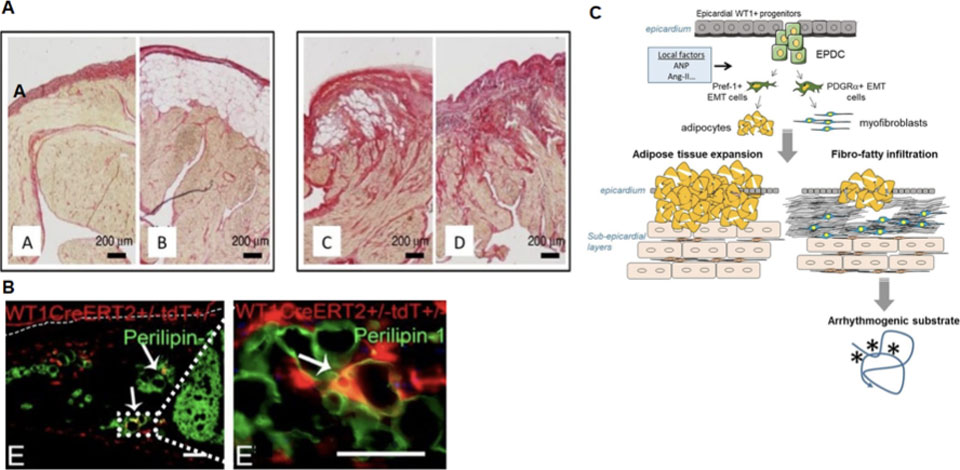
Figure 8. L’épicarde est réactivé précocement au cours de la formation de la cardiomyopathie auriculaire et du substrat de la fibrillation auriculaire. (A) Les composants auriculaires humains tels que le tissu adipeux épicardique (TAE) et la fibrose sont révélés par coloration au rouge Sirius et montrent un épicarde remodelé non fibrotique sans (A) ou avec (B) TAE et un épicarde remodelé fibrotique avec (C) ou sans (D) TAE. (B) Les cellules progénitrices épicardiques (EPDC) sont la source des adipocytes, comme le révèle le modèle murin de traçage de lignée cellulaire. Co-immunomarquage pour la périlipine-1 (marqueur adipogénique) et le WT1-tomato (marquant les EPDC) dans les cellules auriculaires de souris nourries avec un régime riche en graisses. Les flèches indiquent les adipocytes dérivés de l’épicarde co-exprimant le WT1-tomato et la périlipine. (C) Le recrutement des EPDC et leur pré-engagement dans des lignées adipocytaires ou fibroblastiques distinctes peut entraîner une infiltration fibro-adipeuse arythmogène des couches sous-épicardiques auriculaires.
1/ Pour étudier les mécanismes impliqués dans le remodelage du TAE vers la fibrose, nous avons utilisé un régime obésogène chez des souris qui a induit une cardiomyopathie atriale (CMA) caractérisée par un changement du métabolisme auriculaire en faveur du stockage des acides gras à longue chaîne médié par l’activation de la voie de β-oxydation, une vulnérabilité à la FA et une réponse inflammatoire dans les oreillettes (Suffee et al CVR 2022). Ce modèle murin de CMA mime le remodelage TAE-fibrose observé dans les biopsies atriales de patients(figure 9A). Les outils transcriptomiques et de modèles murin transgéniques ont révélé des sous-ensembles de macrophages nichés dans la couche sous-épicardique (Crepin et al bioRxiv 2026) (Figures 9B,C).
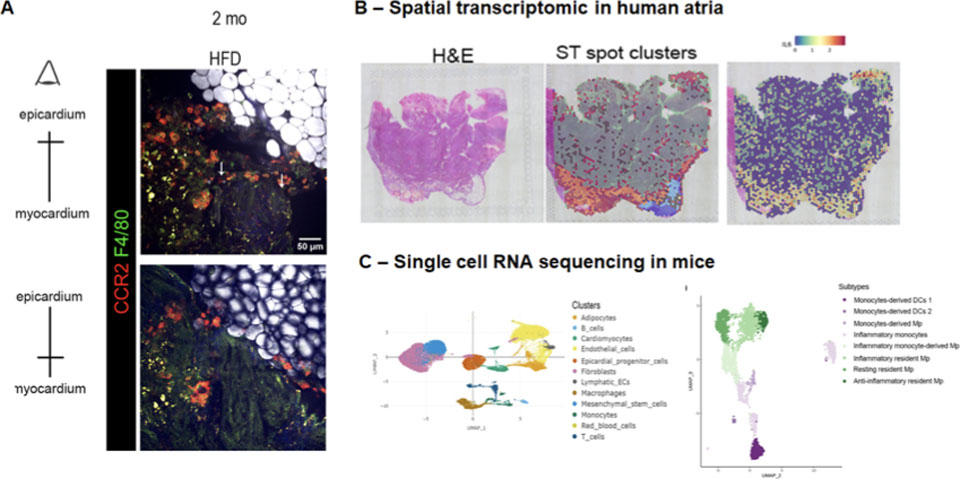
Figure 9. Le modèle murin de cardiomyopathie auriculaire a révélé une infiltration de cellules immunitaires dans l’épicarde. (A) Le modèle murin de cardiomyopathie auriculaire induite par un régime riche en graisses mime le remodelage auriculaire avec accumulation de tissu adipeux, fibrose et infiltration de cellules immunitaires. Des monocytes (CCR2) et les macrophages (F4/80) sont révélées par microscopies spectrale Raman et biphotonique. Les outils transcriptomiques spatiale pour les oreillettes de patients (B) et unicellulaire des oreillettes de souris atteintes de CMA (C) ont révélé des sous-ensembles de macrophages spécifiquement localisés dans le sous-épicarde pathologique.
2/ En collaboration avec N. Farès (Université Saint-Joseph de Beyrouth), nous avons récemment montré que l’augmentation de la disponibilité du peptide natriurétique auriculaire (ANP) myocardique améliore l’insuffisance cardiaque dans la cardiomyopathie diabétique. Nous avons montré que le mANP, un ANP muté résistant à la dégradation par la néprilysine, réduit la dysfonction ventriculaire gauche et la fibrose cardiaque chez les rats diabétiques de type 2 avec une efficacité supérieure à celle de l’ANP. La forme mutée de l’ANP a produit des niveaux plus élevés de GMP cyclique cardiaque et une signalisation anti-fibrotique plus forte in vivo que l’ANP non muté. Par conséquent, la biodisponibilité de l’ANP myocardique est cruciale pour limiter la fibrose et améliorer la fonction cardiaque dans la cardiomyopathie diabétique (Bakhos, Eur Heart J Open, 2025).
Projets en cours
Dans le but d’enrichir les connaissances sur le remodelage TAE vers la fibrose, nos projets s’articulent autour de 4 objectifs :
– La migration des macrophages résidents vers les EPDC suggère une interaction entre les EPDC et les macrophages. À cette fin, nous examinerons l’impact des macrophages sur la différenciation des EPDC.
– Nous nous intéressons également à l’effet des macrophages polarisés sur le phénotype du TAE.
– Dans le modèle murin de CMA, les lymphocytes s’infiltrent dès le premier stade de la maladie, comme observé chez les patients atteints de primo-FA , qu’ils soient obèses ou non. Nous étudions leur rôle dans l’exacerbation du remodelage, notamment par le biais de la réponse inflammatoire.
– La réponse immunitaire est observée dans les maladies arythmogènes tant au niveau des oreillettes que des ventricules. Nous identifions le profil immunitaire et le comparerons afin d’établir un atlas immunitaire.
Outils
Nous utilisons des biopsies auriculaires humaines et des échantillons sanguins pour étudier le phénotype et la fonction des cellules immunitaires que nous associons à l’imagerie, à la cytométrie en flux, à la transcriptomique spatiale et aux outils métabolomiques.
Nous utilisons un modèle de souris transgénique pour le traçage de la lignée des cellules précurseurs et des sous-ensembles de macrophages, et un second modèle présentant une déficience pour des sous-ensembles spécifiques de macrophages. Nous effectuons des mesures cardiaques cliniques et des tests de vulnérabilité à la FA. Pour induire une CMA chronique, nous utilisons un régime obésogène avec une alimentation enrichie en graisses. Pour étudier la CMA aigüe, nous utilisons un modèle d’insuffisance cardiaque avec fraction d’éjection réduite (HFrEF). Pour établir l’analyse du profil des cellules précurseurs et immunitaires, nous utilisons des tests immunologiques, le séquençage d’ARN unicellulaire, le western blot et la cytométrie en flux. Pour tester les stratégies thérapeutiques, nous utilisons des anticorps neutralisants injectés dans des modèles murins de CMA.
Collaborations
Collaborations Internationales :
Indian Institutes of Technology, Delhi, India, I. GUPTA
CHUV, Département Cœur-Vaisseaux, Lausanne, Suisse, N. ROSENBLATT-VELIN
MAESTRIA H2020 Consortium Européen
Cardiovascular Research Center Massachusetts General Hospital, Boston, USA
Université Saint Joseph, Département de physiologie, Beyrouth, Liban, N. FARES
Mayo Clinic College of Medicine, Rochester, USA, J. BURNET
Collaborations Nationales :
Institut Mondor de recherche Biomédicale, Créteil, A. BOISSONAS
Cardiac and Thoracic Surgery, APHP, Paris, G. LEBRETON, P. LEPRINCE, E. BERG, E. GANDJBAKHCH, T. ROLLAND.
Gustave Roussy, Villejuif, S. DURAND
Institut Pasteur, Paris, E. KARKENI
Autorité de Sûreté Nucléaire et de Radioprotection ASNR, Fontenay aux Roses, V. MONCEAU
INSERM U1046 – UMR CNRS 9214 – Université de Montpellier R. ANDRIANTSITOHAINA
CHU Cardiac and Thoracic Surgery, Lille, S. NINI
PARCC, Paris Centre de recherche cardiovasculaire, Paris, H. AIT-OUFELLA
Nutriomics, Paris, E.L. GAUTIER
THEME 4 – A et physiopathologie Cardiovasculaire – Stéphane HATEM
Membres du groupe
Stéphane HATEM, MD, PhD, PUPH
Co direction avec Maharajah PONNAIAH responsable de la plateforme ICAN I/O
Ali RAMMAL, Doctorant
Hind SAHABEDDINE, Doctorante
Kimia SADRADDINI, Doctorant
Contexte scientifique et contributions passées
L’intelligence artificielle (IA) est en train de transformer le paysage des sciences biomédicales et, plus particulièrement, celui de la recherche cardiovasculaire. En effet, l’IA offre aux chercheurs la capacité unique d’analyser des ensembles de données complexes, d’identifier de nouvelles trajectoires ou de nouveaux groupes de données, ce qui permet de mettre en évidence de nouveaux processus pathologiques, de nouvelles sous-populations de patients ou de nouveaux facteurs de risque.
Projets en cours
1/ Approche d’apprentissage automatique pour identifier des groupes de patients présentant une réponse chronotrope cardiaque distincte pendant l’exercice
Nous développons un algorithme d’apprentissage automatique pour analyser la réponse chronotrope cardiaque à un exercice progressif. Nous utilisons un regroupement hiérarchique non supervisé basé sur l’IA et une analyse algorithmique de la trajectoire de la fréquence cardiaque pour identifier des sous-populations de patients atteints de cardiopathies et d’insuffisance cardiaque. Ensuite, à l’aide de différentes approches physiologiques, nous déchiffrons les mécanismes physiopathologiques sous-jacents au regroupement des patients.
2/ Modèles d’apprentissage automatique de l’échocardiographie et de l’imagerie par IRM
Nous avons développé un outil algorithmique combinant l’imagerie cardiaque par résonance magnétique et des méthodes d’apprentissage automatique afin de mettre au point de nouveaux biomarqueurs permettant d’améliorer le diagnostic de la fibrillation atriale. Nous avons mesuré les caractéristiques du tissu adipeux du sillon auriculo-ventriculaire lors d’une IRM cardiaque de routine et avons prouvé qu’il pouvait être utilisé comme indicateur des dépôts de tissu adipeux épicardique auriculaire. Cela a été intégré dans un biomarqueur IRM cardiaque multiparamétrique pour l’identification précoce de la cardiomyopathie auriculaire (Bialobroda, Eur Heart J Imaging Methods Pract, 2024). Il s’agit d’une avancée novatrice dans l’analyse de l’imagerie cardiaque à l’aide de l’IA. Nous avions précédemment utilisé l’IRM pour quantifier différents composants du myocarde de l’oreillette gauche et nous avons pu corréler la fibrose totale, interstitielle et graisseuse avec l’histologie 2D (Bouazizi, Plos One, 2018).
Outils
– Grands ensembles de données (echo CPET) obtenus à partir de cohortes de patients avec suivi clinique
– Infrastructure informatique pour le stockage, l’exploration et l’analyse des données
– Expertise en IA (ICAN I/O, SCAI-SU et SU Abu Dhabi)
Collaborations
Collaborations Internationales :
Mayo Clinic College of Medicine, Rochester, USA, J. BURNET
Collaborations Nationales :
Laboratoire d’Imagerie Biomédicale, U 1146, Paris, A. REDHEUIL

| Name | Position | ORCID |
|---|
- Bakhos, JJ, Saliba, Y, Hajal, J, Achkouty, G, Oskaridjian, H, Albuquerque, M et al.. Inhibiting atrial natriuretic peptide clearance reduces myocardial fibrosis and improves cardiac function in diabetic rats. Eur Heart J Open. 2025;5 (2):oeaf031. doi: 10.1093/ehjopen/oeaf031. PubMed PMID:40201591 PubMed Central PMC11977460.
- Bialobroda, J, Bouazizi, K, Ponnaiah, M, Kachenoura, N, Charpentier, E, Zarai, M et al.. The epicardial adipose tissue confined in the atrioventricular groove can be used to assess atrial adipose tissue and atrial dysfunction in cardiac magnetic resonance imaging. Eur Heart J Imaging Methods Pract. 2024;2 (1):qyae057. doi: 10.1093/ehjimp/qyae057. PubMed PMID:39224099 PubMed Central PMC11367945.
- Bignard, J, Atassi, F, Claude, O, Ghigna, MR, Mougenot, N, Abdoulkarim, BS et al.. T-cell dysregulation and inflammatory process in Gcn2 (Eif2ak4-/-)-deficient rats in basal and stress conditions. Am J Physiol Lung Cell Mol Physiol. 2023;324 (5):L609-L624. doi: 10.1152/ajplung.00460.2021. PubMed PMID:36852942 .
- Solinc, J, Raimbault-Machado, J, Dierick, F, El Bernoussi, L, Tu, L, Thuillet, R et al.. Platelet-Derived Growth Factor Receptor Type α Activation Drives Pulmonary Vascular Remodeling Via Progenitor Cell Proliferation and Induces Pulmonary Hypertension. J Am Heart Assoc. 2022;11 (7):e023021. doi: 10.1161/JAHA.121.023021. PubMed PMID:35348002 PubMed Central PMC9075467.
- Suffee, N, Baptista, E, Piquereau, J, Ponnaiah, M, Doisne, N, Ichou, F et al.. Impacts of a high-fat diet on the metabolic profile and the phenotype of atrial myocardium in mice. Cardiovasc Res. 2022;118 (15):3126-3139. doi: 10.1093/cvr/cvab367. PubMed PMID:34971360 .
- Melgari, D, Barbier, C, Dilanian, G, Rücker-Martin, C, Doisne, N, Coulombe, A et al.. Microtubule polymerization state and clathrin-dependent internalization regulate dynamics of cardiac potassium channel: Microtubule and clathrin control of KV1.5 channel. J Mol Cell Cardiol. 2020;144 :127-139. doi: 10.1016/j.yjmcc.2020.05.004. PubMed PMID:32445844 .
- Suffee, N, Moore-Morris, T, Jagla, B, Mougenot, N, Dilanian, G, Berthet, M et al.. Reactivation of the Epicardium at the Origin of Myocardial Fibro-Fatty Infiltration During the Atrial Cardiomyopathy. Circ Res. 2020;126 (10):1330-1342. doi: 10.1161/CIRCRESAHA.119.316251. PubMed PMID:32175811 .
- Eyries, M, Montani, D, Girerd, B, Favrolt, N, Riou, M, Faivre, L et al.. Familial pulmonary arterial hypertension by KDR heterozygous loss of function. Eur Respir J. 2020;55 (4):. doi: 10.1183/13993003.02165-2019. PubMed PMID:31980491 .
- Beuriot, A, Eichel, CA, Dilanian, G, Louault, F, Melgari, D, Doisne, N et al.. Distinct calcium/calmodulin-dependent serine protein kinase domains control cardiac sodium channel membrane expression and focal adhesion anchoring. Heart Rhythm. 2020;17 (5 Pt A):786-794. doi: 10.1016/j.hrthm.2019.12.019. PubMed PMID:31904424 .
- Keck, M, Flamant, M, Mougenot, N, Favier, S, Atassi, F, Barbier, C et al.. Cardiac inflammatory CD11b/c cells exert a protective role in hypertrophied cardiomyocyte by promoting TNFR2- and Orai3- dependent signaling. Sci Rep. 2019;9 (1):6047. doi: 10.1038/s41598-019-42452-y. PubMed PMID:30988334 PubMed Central PMC6465256.
- Eyries, M, Montani, D, Nadaud, S, Girerd, B, Levy, M, Bourdin, A et al.. Widening the landscape of heritable pulmonary hypertension mutations in paediatric and adult cases. Eur Respir J. 2019;53 (3):. doi: 10.1183/13993003.01371-2018. PubMed PMID:30578383 .
- Launay, T, Momken, I, Carreira, S, Mougenot, N, Zhou, XL, De Koning, L et al.. Acceleration-based training: A new mode of training in senescent rats improving performance and left ventricular and muscle functions. Exp Gerontol. 2017;95 :71-76. doi: 10.1016/j.exger.2017.05.002. PubMed PMID:28479388 .
- Suffee, N, Moore-Morris, T, Farahmand, P, Rücker-Martin, C, Dilanian, G, Fradet, M et al.. Atrial natriuretic peptide regulates adipose tissue accumulation in adult atria. Proc Natl Acad Sci U S A. 2017;114 (5):E771-E780. doi: 10.1073/pnas.1610968114. PubMed PMID:28096344 PubMed Central PMC5293064.
- Eichel, CA, Beuriot, A, Chevalier, MY, Rougier, JS, Louault, F, Dilanian, G et al.. Lateral Membrane-Specific MAGUK CASK Down-Regulates NaV1.5 Channel in Cardiac Myocytes. Circ Res. 2016;119 (4):544-56. doi: 10.1161/CIRCRESAHA.116.309254. PubMed PMID:27364017 .
- Dierick, F, Héry, T, Hoareau-Coudert, B, Mougenot, N, Monceau, V, Claude, C et al.. Resident PW1+ Progenitor Cells Participate in Vascular Remodeling During Pulmonary Arterial Hypertension. Circ Res. 2016;118 (5):822-33. doi: 10.1161/CIRCRESAHA.115.307035. PubMed PMID:26838788 .
- Haemers, P, Hamdi, H, Guedj, K, Suffee, N, Farahmand, P, Popovic, N et al.. Atrial fibrillation is associated with the fibrotic remodelling of adipose tissue in the subepicardium of human and sheep atria. Eur Heart J. 2017;38 (1):53-61. doi: 10.1093/eurheartj/ehv625. PubMed PMID:26612579 .
- Venteclef, N, Guglielmi, V, Balse, E, Gaborit, B, Cotillard, A, Atassi, F et al.. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo-fibrokines. Eur Heart J. 2015;36 (13):795-805a. doi: 10.1093/eurheartj/eht099. PubMed PMID:23525094 .
- Germain, M, Eyries, M, Montani, D, Poirier, O, Girerd, B, Dorfmüller, P et al.. Genome-wide association analysis identifies a susceptibility locus for pulmonary arterial hypertension. Nat Genet. 2013;45 (5):518-21. doi: 10.1038/ng.2581. PubMed PMID:23502781 PubMed Central PMC3983781.
Blandin C, Dilanian D, Fontaine V, Mougenot N, Gravez B, Bobin P, Duboscq-Bidot L, Farhi D, Chardonnet S, Nadaud S, Sanchez-Alonzo JL, Shevchuk A, Gorelik J, Gandjbakhch E, Hatem SN, Villard E, Balse E. Depleting trafficking regulator CASK promotes intercalated disc organization and ventricular function. BioRxiv. 2024. doi.org/10.1101/2024.10.14.618172
Nadine Suffee
Prix Alain Castaigne – 2023
Stéphane Hatem
Prix Alain Castaigne – 2018
Florent Soubrier
Prix Jean Valade – 2014
Prix Lamonica de Cardiologie – Académie des Sciences – 2017
France Diérick
Prix Marion Elizabeth Brancher – 2017

