TEAM 1 – Philippe Charron – Genomics and Pathophysiology of Myocardial Diseases: from Monogenic to Complex Diseases

Philippe Charron, Team 1 Head
CLINICAL AND SCIENTIFIC CONTEXT OF THE RESEARCH PROJECT
Cardiomyopathies and channelopathies, the two major subgroups of hereditary cardiac diseases, are the principal causes of sudden cardiac death and heart failure in young patients (<40 years), especially in athletes. Despite improvements in the care of these diseases, thanks to pharmacological drugs, implantable cardioverter defibrillators and cardiac transplantation, new knowledge on the underlying genetic causes, on metabolic pathways and physiopathology are needed to identify new targets or therapeutic strategies and to better prevent the devastating complications of these pathologies.
In addition, recent advances in the understanding of the diseases’ causes (in particular in the sarcomeric and ionic canal genes’ variants) led to a new comprehension of the complex interaction between genetic architecture (rare, mutation, and frequent variants), environmental factors (sport, myocarditis, drugs) or patients’ gender. These new knowledge and new paradigm have important consequences on the global understanding of the sarcomeric and ionic canal proteins’ physiology as well as on the physiopathology of complex diseases such as heart failure and arrythmia.
Since more than 20 years, our group is interested in deciphering the genetic and cellular mechanisms underlying the development of cardiomyopathies and channelopathies. We recently identified new rare and frequent variants implied in these diseases through genomic strategies of association analyses (GWAS) or sequencing. We study underlying signaling pathways and initiate new therapeutic strategies based on these scientific advances. Simultaneously, we develop, in clinical practice, translational approaches implying genetic testing and high throughput resequencing, to improve medical care of the patients and their families thanks to personalized medicine.
Team 1 develops research strategies around 4 axes:
- Transfer new knowledge on genetics and physiopathology in clinical practice, notably through genetic testing, and high throughput resequencing, to develop personalized medicine
- Identify key-actors (rare and frequent genetic variants, signaling pathways) in cardiomyopathies and channelopathies via multiomics approaches
- Understanding the functions, interactions and physiopathology of the principal actors of these diseases through human cardiomyocytes derived from induced pluripotent stem cells (iPSC) and murine models.
- Progress towards new therapies (pharmacological, interventional, genetics) in monogenic and/or complex forms of heart failure and arrythmia.
Team 1 is member of Ecole doctorale 394, Physiologie, Physiopathologie et Thérapeutique.
THEME 1 – GENOMICS OF HEART FAILURE DUE TO DILATED CARDIOMYOPATHY
Eric Villard, Sophie Garnier, Richard Isnard, Philippe Charron
Background. Dilated cardiomyopathy is a complex disease with both familial and sporadic formas. Unraveling the gentic component of this disease to improve patients’ care and find nex therapeutic targets is one on the leading task of our group. In familial forms, the linkage analysis/positional candidate gene strategy has been developed (Sylvius N, 2001), completed now by next generation exome and genome sequencing. for sporadic forms, the candidate gene strategy was quite unfruitful and, rapidly, we turned to high throughput association studies on thousands of cases and controls. Successive GWAS on pooled DNA samples (Villard E, 2011) or using exomic enriched SNPs (Esslinger U, 2017) revealed 8 candidate genomic loci (HSPB7, TTN, SLC39A8, MLIP, FLNC, BAG3, ALPK3 and FHOD3), two being also implied in Ischemic Heart Failure (Garnier S, 2015). A third GWAS on 2,700 sporadic DCM cases and 4,400 controls, combined with an imputation analysis, is now ongoing to identify new susceptibility loci.
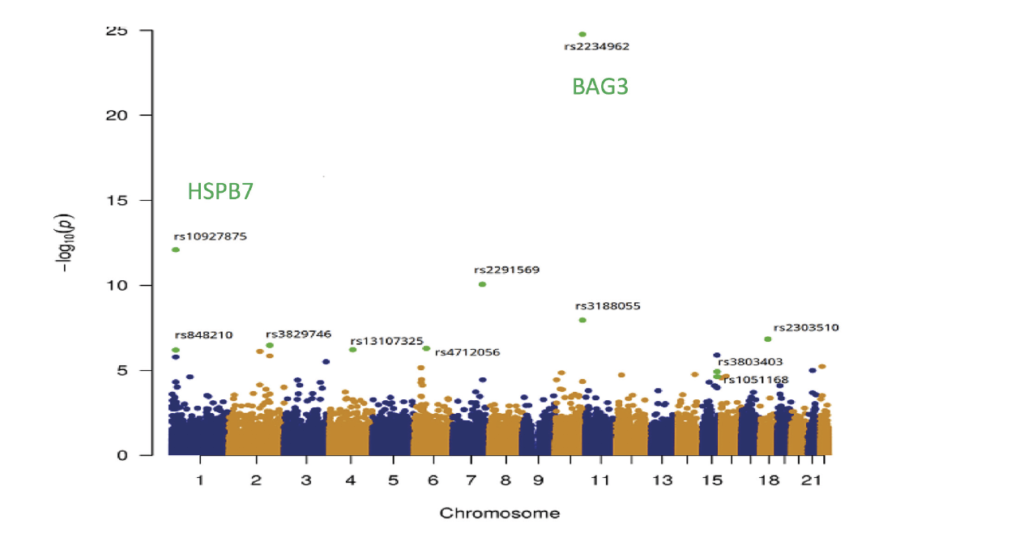
Manhattan Plot – Exome Wide Association Study. 95.499 variants were investigated by logistic regression analysis. Associations are summarized in the Manhattan plot which displays (green dots), the eleven SNVs significantly associated with DMC. From Esslinger U and al. 2017.
Our group is also deeply implied in the pathphysiological impact of the discovered genes. Based on our results from GWAS/EWAS in DCM we hypothesize that prtein quality control, in the context of repeated mechanical stress, is a major pathway for cardiomyocytes health. Such hypotheses, and potential therapeutic applications, are questioned using knock-in mouse models of DCM and iPs-derived cardimyocytes with Engeniered Heart tissue (EHT) cutured from mutated patient’s cells. We also initiated a collaborative project with pharmaceutical industry whose purpose is to identify the molecular pathways implicated in hyoertrophic cardio myopathy and perform molecular screen based on iPs-derived cardiomyocytes. We take advantages from our strong anchoring within the national referral centre for cardiac hereditary diseases and unique access to the patients and their genotypes, the in-house facility dedicated to iPs-cardiomyocytes and our skills in genomic editing.
THEME 2 – Genomic analysis of abnormal cardiac repolarization response to pharmacologic drugs
Joe-Elie Salem, Christian Funck-Brentano
Many drugs used for non-cardiovascular and cardiovascular purposes, such as sotalol, have the side effect of prolonging cardiac repolarization, which can trigger life-threatening cardiac arrhythmias by inhibiting the potassium-channel IKr (KCNH2). ECG changes (such as QTc, Tpeak-Tend, Tamp, T wave notches) vary markedly between subjects, suggesting the existence of predisposing genetic factors. In the GENEREPOL study, 990 healthy individuals, prospectively challenged with an oral sotalol dose, were monitored for changes in ventricular repolarization on ECG. QTc and TpTe increased and TAmp decreased. A random subsample of 489 individuals were subjected to a genome-wide-association analysis where 8,306,856 imputed single nucleotide polymorphisms (SNPs) were tested for association with QTc, TpTe and TAmp modulations (D), as well as their derived principal-components, and notches apparition. None of the studied SNPs reached the statistical threshold (Salem JE et al, 2017). The study, however, showed that a principal component analysis based on DQTcF, DTAmp and DTpTe might be an integrative way to further differentiate patients with the most extreme IKr inhibition. We patented this diagnostic algorithm in 2016 as a method of determining the susceptibility for induction of a torsade de pointes (PCT/EP2017/058714, BRV 116 – WO — 2016-096).
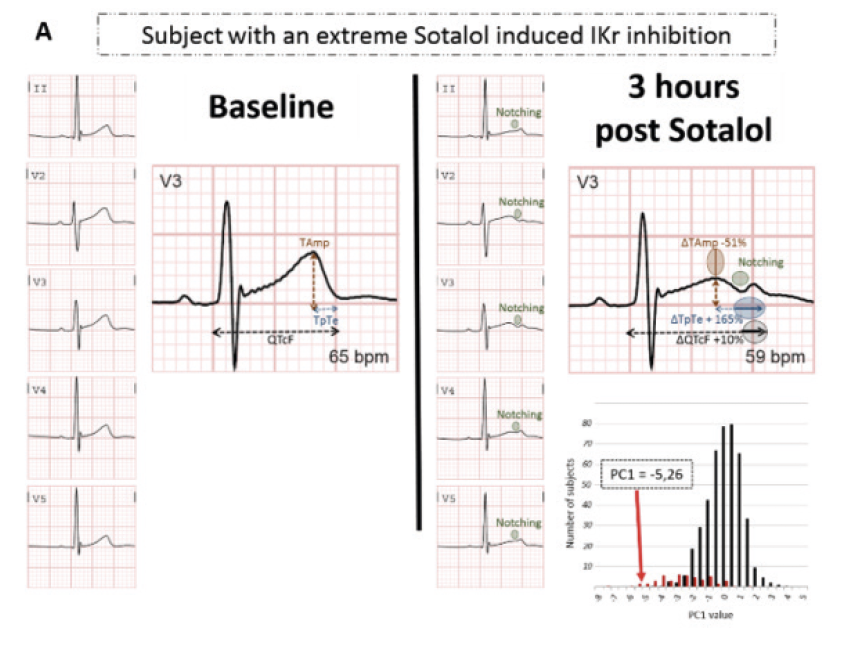
The PC1 values and distribution of dicovery cohort are shown. PC1 is issued from principal component analysis of ΔQTc, ΔTpTe and ΔTAmp. “Notcher” subjects are represented in red and “non notcher” in black. Typical QT and t-ware changes (A) in a subject with a pronounced sotalol-induced IKr inhibition indicated by a notch and (B) in a subject with subject with minimal sotalol-induced IKr inhibition.
Our current project is to understand the determinants of gender specific repolarization. Cardiac repolarization is influenced by complex interactions between sex steroid hormones and gonadotropins (Salem JE et al, 2016), depending on gender. We recently showed that the progesterone/estradiol ratio in women, testosterone in men, and FSH in both genders are major determinants of ventricular repolarization (Abehsira G et al, 2016). We are planning to further study in GENEREPOL cohort the influence of sex hormones on drug induced QT prolongation, combined with the used of whole-exome sequencing seeking for rare and low frequency variants. We will also seek for interaction between hormones and expression of genes using cardiomyocytes derived from iPS cells of patients with congenital or drug-induced long QT syndrome. We plan to test if an appropriate exogenous hormonal administration might increase the repolarization reserve in situation at risk. We already patented a method of treatment of “torsades de pointes” by exogenous hormonal administration (PCT/EP2017/059097, BRV 119 – WO — 2016-120).
THEME 3 – From deciphering the genetic determinant of ventricular arrhythmia to therapeutic applications
Nathalie Neyroud, Fabrice Extramiana, Antoine Leenhardt, Pascale Guicheney
Our group has been interested for many years in deciphering the genetic background of cardiac channelopathies. We study functional consequences of new mutations in ion-channel subunits and associated proteins with the aim of increasing our understanding of regulatory processes of ion channels involved in human cardiac pathophysiology. We use molecular, immunohistochemical and electrophysiological techniques, associated to viral gene transfer and animal and cellular models to identify new genetic variants and to determine how variants linked to arrhythmia can alter the expression and function of cardiac voltage-gated channels.
The SCN5A gene encodes the human cardiac sodium channel α-subunit, Nav1.5, responsible for initiation and propagation of the action potential. Loss-of-function mutations in SCN5A have been linked to life-threatening cardiac arrhythmias (such as the Brugada syndrome or sick sinus syndrome). Our projects center on several models to study SCN5A deficiency: 1) a murine model of Scn5a haplo-insufficiency, partially recapitulating the Brugada syndrome phenotype. Our group seeks to overexpress the cardiac sodium channel in Scn5a deficient mice using viral vectors in an attempt to restore their sodium current and ECG parameters (see Figure below). 2) a human model of iPS (induced pluripotent stem) cells, the genome of which has been edited to induce an heterozygous deficiency of SCN5A. New therapeutical approaches are tested on these different models.
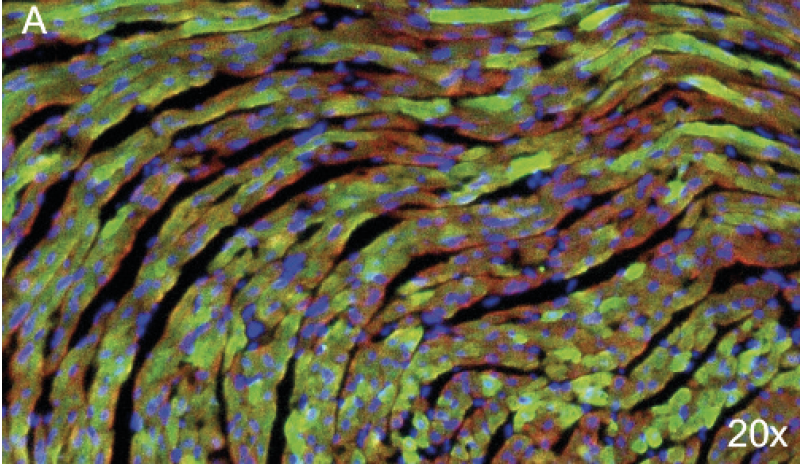
Eight-micrometer slices of heart tissue from a mouse injected with the AAV carrying SCN5A. AAV injection led to the expression of Nav1.5 tagged with the GFP in almost 50% of heart cells. A. Nuclei are stained in blue, a-actinin in red and the GFP in green (20x).
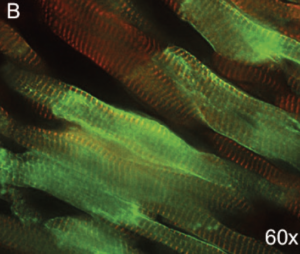
Magnification of transduced cardiomyocytes (60x).
Idiopathic Ventricular Fibrillation (IVF) is a rare cause of sudden cardiac arrest. The exact incidence of IVF is unknown but is declining with the advance of diagnostic testing and the discovery of primary arrhythmia syndromes. For more than 15 years, our group has worked to investigate patients presenting with ventricular tachycardia and to collect their familial information and DNA. We have recently selected a group of cases diagnosed with IVF originating from Purkinje fibers. Our project seeks to identify new genes and variants involved in IVF by genomic approaches.
THEME 4 – Identify new physiological pathways in Arrhythmogenic right ventricular cardiomyopathy using innovative human cellular models
Estelle Gandjbakhch, Eric Villard
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a rare inherited cardiomyopathy characterized by fibrofatty replacement of myocytes leading to ventricular arrhythmia, sudden death and heart failure mainly caused by desmomosal genes mutations (Fressard V, 2010). Ongoing work by our team has shown the essential role of cadherin mediated adhesion in the disease (Vite A,2013 & Vite A., submitted). One major challenge in translational research of ARVC is to generate a pertinent cardiac cellular model expressing mature desmosomes and recapitulating cyclic mechanical load. In this project, we will generate an IPS-derived engineered heart tissue (hIPS-EHT) which will be used as an innovative in vitro model to unravel physiopathology of ARVC and explain the mutation specific phenotypes. This 3D cardiac model reconstitutes a mature contractile cardiac tissue reproducing mechanical stress, as observed in vivo. We aim to structurally compare the effect of PKP2 and DSG2 mutations on cardiomyocyte and junction structures using 3D immunofluorescent imaging as well as their consequences on desmosome function and cardiomyocytes electrophysiological properties. We also aim to study the expression profiles associated with PKP2 and DSG2 mutations by whole-transcriptome analysis using high throughput RNA sequencing.
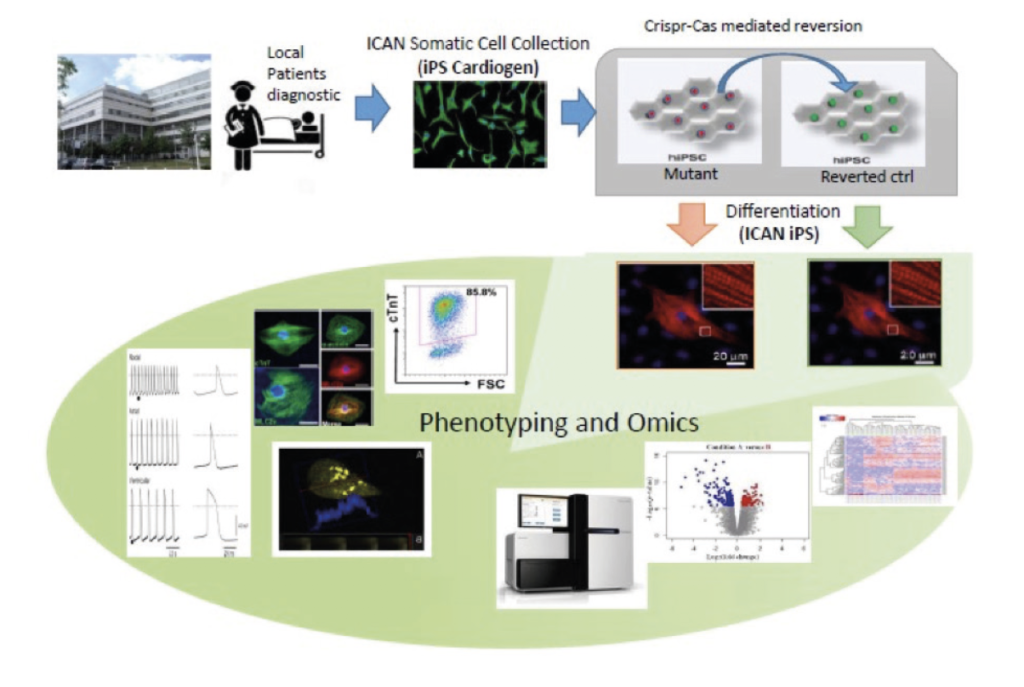
Production of human cardiomyocytes derived from patient iPS cells with mutation of interest
THEME 5 – Translational impact of genetic testing on precision medicine
Pascale Richard, Véronique Fressart, Estelle Gandjbakhch, Philippe Charron
Apart from the use of genetic results in the cascade screening of families with hereditary diseases (Charron P, 2010 & Cirino AL, 2017), our team has reported several publications about the role of some genes or mutations in the severity of cardiac diseases, such as the role of multiple mutations on the prognosis of a cardiomyopathy (Richard P, 2002, Fressard V, 2010) or the role of some particular genes such as PRKAG2 & GLA in HCM (Thevenon J, 2016, Séné T, 2016) or DSG2 in ARVC (Hermida A, in press). Identification of these genetic factors may help to identify high risk profile patients that would benefit from close cardiac monitoring and early treatments such as implantable cardioverter defibrillator or heart failure therapy. An illustrative example of direct personalized therapeutic impact is about defibrillator implantation decision based on genetics. Our team actively participated to international joint efforts to establish the independent prognostic role of LMNA mutation, associated with high risk of sudden death, and to clarify the identification of patients that would benefit most from implantable cardiac defibrillator implantation (van Rijsingen IA, 2012; Kumar S, 2016; Thuillot M, 2019).
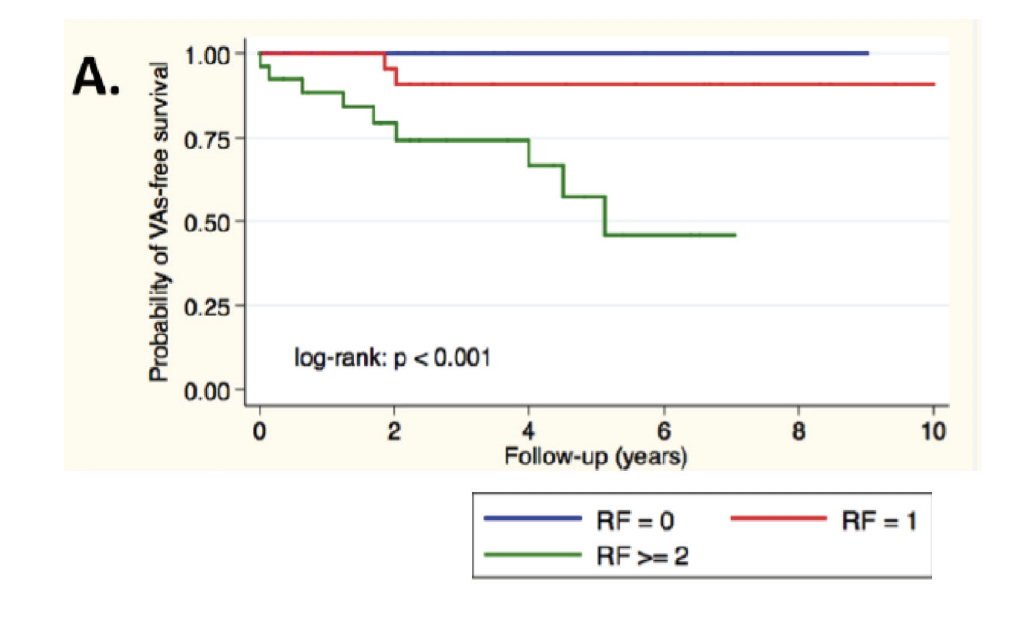
First validation of a prognostic score in heart failure due to LMNA. Four predicitve factors (non sustained ventricular tachycardia. LV Ejection Fraction <45%, male sex and non-missense mutations) for malignant ventricular arrythmia (VA) were studied in a monocentric cohort. ICD is recommended in the presence of 2 risk factors. From Thuillot M. & al., 2019.
Refine the genetic spectrum of cardiomyopathies with high throughput sequencing. Large cohorts of patients with Left ventricle non-compaction (LVNC), HCM and DCM have been collected through the national network of the Referral center for cardiac hereditary diseases. Resequencing strategy with various panels of genes, including a panel of >100 genes involved in various cardiac hereditary diseases are under way in order to refine the prevalence of genes, the overlap between cardiomyopathy subtypes and then progress towards optimal diagnostic strategies for routine care of patients and families.

| Name | Position | ORCID |
|---|
- Hermida, A, Ader, F, Millat, G, Jedraszak, G, Vogel, L, Garçon, L et al.. RBM20 Gene in Patients With Cardiomyopathy: Phenotypic Expression for Loss-of-Function Versus Hotspot Variants. Circ Heart Fail. 2025;18 (3):e012492. doi: 10.1161/CIRCHEARTFAILURE.124.012492. PubMed PMID:39823286 .
- Jurgens, SJ, Rämö, JT, Kramarenko, DR, Wijdeveld, LFJM, Haas, J, Chaffin, MD et al.. Genome-wide association study reveals mechanisms underlying dilated cardiomyopathy and myocardial resilience. Nat Genet. 2024;56 (12):2636-2645. doi: 10.1038/s41588-024-01975-5. PubMed PMID:39572784 PubMed Central PMC11631763.
- Zheng, SL, Henry, A, Cannie, D, Lee, M, Miller, D, McGurk, KA et al.. Genome-wide association analysis provides insights into the molecular etiology of dilated cardiomyopathy. Nat Genet. 2024;56 (12):2646-2658. doi: 10.1038/s41588-024-01952-y. PubMed PMID:39572783 PubMed Central PMC11631752.
- Ader, F, Jedraszak, G, Janin, A, Billon, C, Buisson, NR, Bloch, A et al.. Prevalence and phenotypes associated with ALPK3 null variants in a large French multicentric cohort: Confirming its involvement in hypertrophic cardiomyopathy. Clin Genet. 2024;105 (6):676-682. doi: 10.1111/cge.14505. PubMed PMID:38356193 .
- Ferrand, MC, Giordano, G, Mougenot, N, Laporte, PL, Vignier, N, Leclerc, A et al.. Intracardiac electrophysiology to characterize susceptibility to ventricular arrhythmias in murine models. Front Physiol. 2024;15 :1326663. doi: 10.3389/fphys.2024.1326663. PubMed PMID:38322613 PubMed Central PMC10846502.
- Perret, C, Proust, C, Esslinger, U, Ader, F, Haas, J, Pruny, JF et al.. DNA-pools targeted-sequencing as a robust cost-effective method to detect rare variants: Application to dilated cardiomyopathy genetic diagnosis. Clin Genet. 2024;105 (2):185-189. doi: 10.1111/cge.14427. PubMed PMID:37904629 .
- Kato, K, Isbell, HM, Fressart, V, Denjoy, I, Debbiche, A, Itoh, H et al.. Novel CALM3 Variant Causing Calmodulinopathy With Variable Expressivity in a 4-Generation Family. Circ Arrhythm Electrophysiol. 2022;15 (3):e010572. doi: 10.1161/CIRCEP.121.010572. PubMed PMID:35225649 .
- Gizon, M, Duboscq-Bidot, L, El Kassar, L, Bobin, P, Ader, F, Giraud-Triboult, K et al.. Generation of a heterozygous SCN5A knockout human induced pluripotent stem cell line by CRISPR/Cas9 edition. Stem Cell Res. 2022;60 :102680. doi: 10.1016/j.scr.2022.102680. PubMed PMID:35093717 .
- Blancard, M, Touat-Hamici, Z, Aguilar-Sanchez, Y, Yin, L, Vaksmann, G, Roux-Buisson, N et al.. A Type 2 Ryanodine Receptor Variant in the Helical Domain 2 Associated with an Impairment of the Adrenergic Response. J Pers Med. 2021;11 (6):. doi: 10.3390/jpm11060579. PubMed PMID:34202968 PubMed Central PMC8235491.
- Doisne, N, Grauso, M, Mougenot, N, Clergue, M, Souil, C, Coulombe, A et al.. In vivo Dominant-Negative Effect of an SCN5A Brugada Syndrome Variant. Front Physiol. 2021;12 :661413. doi: 10.3389/fphys.2021.661413. PubMed PMID:34122134 PubMed Central PMC8195286.
- Garnier, S, Harakalova, M, Weiss, S, Mokry, M, Regitz-Zagrosek, V, Hengstenberg, C et al.. Genome-wide association analysis in dilated cardiomyopathy reveals two new players in systolic heart failure on chromosomes 3p25.1 and 22q11.23. Eur Heart J. 2021;42 (20):2000-2011. doi: 10.1093/eurheartj/ehab030. PubMed PMID:33677556 PubMed Central PMC8139853.
- Touat-Hamici, Z, Blancard, M, Ma, R, Lin, L, Iddir, Y, Denjoy, I et al.. A SPRY1 domain cardiac ryanodine receptor variant associated with short-coupled torsade de pointes. Sci Rep. 2021;11 (1):5243. doi: 10.1038/s41598-021-84373-9. PubMed PMID:33664309 PubMed Central PMC7970841.
- Vite, A, Gandjbakhch, E, Hery, T, Fressart, V, Gary, F, Simon, F et al.. Desmoglein-2 mutations in propeptide cleavage-site causes arrhythmogenic right ventricular cardiomyopathy/dysplasia by impairing extracellular 1-dependent desmosomal interactions upon cellular stress. Europace. 2020;22 (2):320-329. doi: 10.1093/europace/euz329. PubMed PMID:31845994 .
- Richard, P, Ader, F, Roux, M, Donal, E, Eicher, JC, Aoutil, N et al.. Targeted panel sequencing in adult patients with left ventricular non-compaction reveals a large genetic heterogeneity. Clin Genet. 2019;95 (3):356-367. doi: 10.1111/cge.13484. PubMed PMID:30471092 .