


TEAM 3 – Elise Balse and Sophie Nadaud – Molecular and Cellular Plasticity in Cardiovascular Diseases

Elise Balse & Sophie Nadaud, Team 3 Heads
Objectives
Cardiovascular diseases are often the result of profound modifications of functional and structural properties of heart and vessels that are caused by a variety of individual and environmental factors. Different mechanisms at molecular and cellular levels and shared between heart and vessels underly this remodeling process. Our team aims at understanding the drivers of the molecular and cellular plasticity that characterize cardiovascular remodeling in the context of distinct cardiovascular diseases: atrial fibrillation (AF), heart failure (HF), and systemic (HTA) and pulmonary (PH) hypertension. These diseases are leading causes of mortality and morbidity worldwide and share epidemiological characteristics notably in the context of metabolic diseases such diabetes, MAFLD or obesity.
All our projects are supported by the excellent scientific and technologic environment of ICAN Institute (Cardiometabolism and Nutrition Institute). In particular, we are assisted by the scientific and technological core labs of ICAN Institute: ICAN BioCell iPS to obtain iPS-derived cells, ICAN-Omics for metabolomic and lipidomics, ICAN-I/O for mathematical modelling, statistics or artificial intelligence.
Our projects focus on
– the plasticity of cellular composition of cardiovascular tissues. We notably study the capacity of progenitor and stem cells to be recruited, to differentiate in various mesenchymal cell lineages and to contribute to atrial and vascular remodeling.
– the plasticity of macromolecular protein complexes regulating cardiac function and their role in pump dysfunction and arrhythmias. We focus on the regulation of ion channels trafficking and targeting in cardiomyocytes.
– the role of metabolic diseases in regulating vascular and myocardial remodeling.
– the role of natriuretic peptide in the heart failure context in diabetic subjects.
– the role of immune and inflammatory cells during atrial cardiomyopathy.
– the use of artificial intelligence to better understand and predict cardiovascular pathophysiology and identify new patient groups or risk factors.
Passed discoveries
In recent years, our team has identified various mechanisms involved in cardiac or vascular remodeling. We have demonstrated alterations of ion channel dynamics in cardiomyocytes in the context of atrial remodeling and atrial fibrillation. We have identified epicardial progenitor cells that participate in the atrial remodeling leading to atrial fibrillation. We also identified vascular progenitor cells that are activated during pulmonary hypertension leading to vascular remodeling.
Research themes
THEME 1 – Myocyte Organization, Ion Channel Sorting and Targeting – Elise Balse
Group members
Elise BALSE, PhD, University Senior Lecturer
Gilles DILANIAN, Engineer
Dounia FAHRI, PhD student
Athina KOUMANTAROU, PhD student
Scientific context and past contributions
The functional expression of ion channels in the sarcolemma of myocytes determines the shape and duration of the action potential, thereby controlling the effective refractory period of the myocardium. The correct functional expression of ion channels can be disrupted at several levels, including at the transcriptional, translational, and post-translational levels. In past years, we have contributed to a better understanding of the dynamics of surface expression of ion channels in cardiac myocytes, particularly the atria-specific repolarization channel KV1.5 in the context of atrial remodeling and atrial fibrillation (Figure 1) (Balse et al. PNAS 2009, Balse et al., Physiol Rev 2012; Boycott et al., PNAS 2013; Melgari et al., JMCC 2020). We have also studied the function of members of the MAGUK protein family in the regulation of cardiac potassium channels (El-Haou et al., 2009: Abi-Char et al., Amer. J Physiol. 2008) and the cardiac sodium channel (Eichel et al., Circ Res 2016; Beuriot et al. Heart Rhythm 2020) (Figure 2).
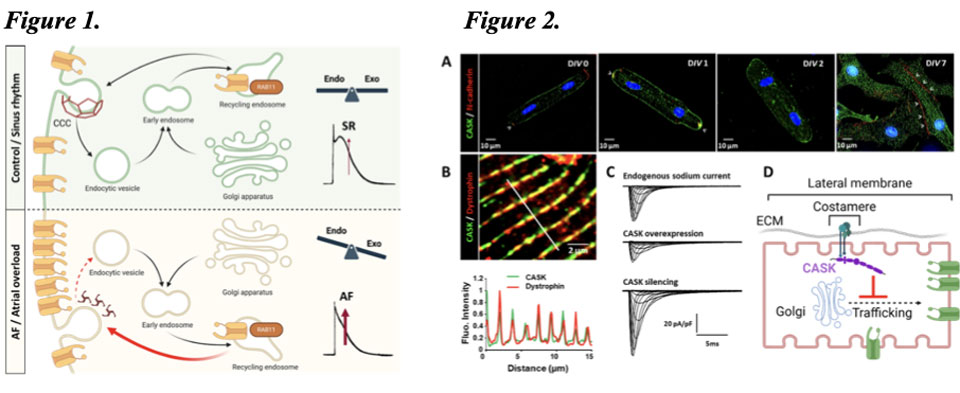
Figure 1. Lability of KV1.5 channel surface expression. (Top) Anterograde and retrograde intracellular trafficking processes are in equilibrium under physiological conditions. (Bottom) In pathological situations, such as chronic atrial hemodynamic overload or atrial fibrillation, exocytosis processes predominate, leading to accumulation of KV1.5 channels on the cardiomyocyte surface and shortening of the atrial action potential. Figure 2. The MAGUK CASK protein, an unconventional partner of the cardiac sodium channel NaV1.5. A) CASK is a protein specifically localized to the lateral membrane, as shown by its exclusion from labeling with the protein N-cadherin. B) CASK co-localizes with dystrophin at the cardiomyocyte costamere. C) Unlike other identified partners, CASK negatively regulates sodium current. D) Summary diagram showing that CASK regulates the functional expression of the channel by acting on its anterograde trafficking
Among MAGUK proteins, we pay particular attention to the protein CASK, which appears to regulate, in addition to the sodium channel, various components of the connexome such as proteins of gap junctions and desmosomes in physiological condition and in the context of Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) (Blandin et al., bioRxiv 2024) (Figure 3).
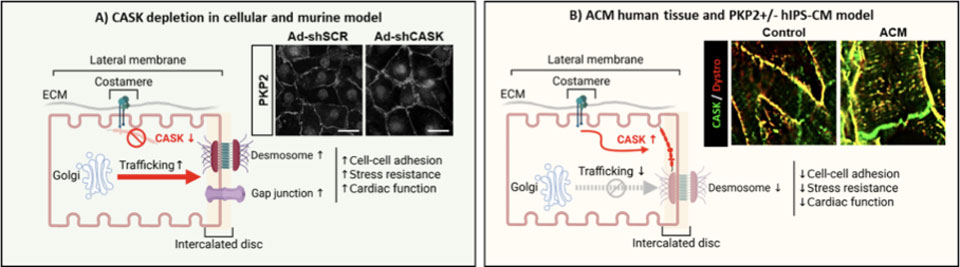
Figure 3. CASK, the controller of connexome protein traffic . A) CASK depletion promotes the transport of connexome proteins (connexin 43 and plakophilin-2) to the intercalated disc in different cell models (neonatal cardiomyocytes, induced pluripotent stem cell-derived cardiomyocytes, hIPS-CM). In a cell model carrying a desmosomal mutation responsible for ARVC (PKP2+/- hIPS-CM), CASK depletion improves cell cohesion and stress resistance. In rats, CASK depletion by AAV vector promotes cardiac function and contractility. B) In biopsies from patients with ARVC , CASK expression is increased and relocalized to the intercalated disc and its.
Ongoing projects
Our work has led us to focus more specifically on the link between electrical properties and the three-dimensional organization of cardiomyocytes, particularly membrane micro-domains, in physiological and pathophysiological contexts. Our current work is divided into two lines of research:
– Determining the role of lateral membrane signaling, and more specifically the CASK protein, in the organization and maintenance of the intercalated disc during postnatal cardiac development and in the context of ARVC.
– Identifying developmental alterations in cardiomyocyte structure induced by a maternal obesogenic diet leading to heart failure in the offspring in adulthood.
Tools
We use mouse models (rats, mice) for a variety of functional studies (Doppler echocardiography and strain echocardiography, pressure-volume relationship, ECG) to study the effects of a CASK invalidation strategy using AAV vectors and the cardiac function of offspring born to obese mothers following a high-fat diet. We use in vitro cell models ranging from primary cardiomyocyte cultures to hIPS-derived cells (cardiomyocytes, epicardial progenitors), both healthy and carrying causal mutations for ARVC. In these in vitro models, we use adenoviral vectors to overexpress or invalidate the expression of proteins of interest. Our expertise includes classical electrophysiology (patch-clamp), high-resolution molecular and tissue imaging. Our research also relies on omics approaches (RNAseq, proteomics, metabolomics, lipidomics) and collaborations with clinicians at the IHU ICAN.
Significance
Our research aims to better understand the mechanisms of protein trafficking and targeting in membrane microdomains in cardiac myocytes in order to highlight cardiac plasticity reserves in hereditary diseases such as ARVC, or acquired diseases such as congenital heart failure.
Collaborations
International collaborations:
Imperial College London, J. GORELIK
National collaborations:
Institut Pasteur Paris, T. WEI
I2MC, Toulouse, C. GALES
THEME 2 – Cellular and Molecular Plasticity During Vascular Remodeling in Arterial Hypertension, Pulmonary Arterial Hypertension and Pulmonary Veno-Occlusive Disease Development – Sophie NADAUD & Florent SOUBRIER
Group members
Sophie NADAUD, PhD, INSERM Researcher
Florent SOUBRIER, MD, PhD, Professor Emeritus
Fabrice ATASSI, Engineer
Thin Hinan NABET, PhD student
Erika LAURO, PhD student
Lylia HAKEM, Master student
Ritej MAHJOUB, Master student
Scientific context and past contributions
Our group is interested in the cellular and molecular factors involved in regulating vascular remodeling. The structural alterations of small vessels (mainly arteries) are major players in the development of pulmonary and systemic hypertension.
Pulmonary hypertension:
1/ We have investigated the origin of new smooth muscle cells produced during pulmonary arterial hypertension. This is a rare and devastating disease with no curative options, characterized by an occlusive remodeling of the distal pulmonary vasculature that ultimately leads to right heart failure. Non-muscularized vessels become muscularized and vessels media thickness is increased with in addition, neointima formation. Resident pulmonary progenitors participate in the pulmonary hypertension-associated vascular remodeling by generating new smooth muscle cells. We have identified resident CD34+/PW1+/PDGFRα+ progenitors involved in the early neomuscularization observed during chronic hypoxia (CH: a model for moderate pulmonary hypertension) (Dierick, Circ Res 2016, Bordenave, ATVB 2020). We have recently demonstrated that the proliferation of these progenitor cells is under the control of the PDGFRα pathway (Solinc, JAHA 2022). Figure 4, 5, 6.
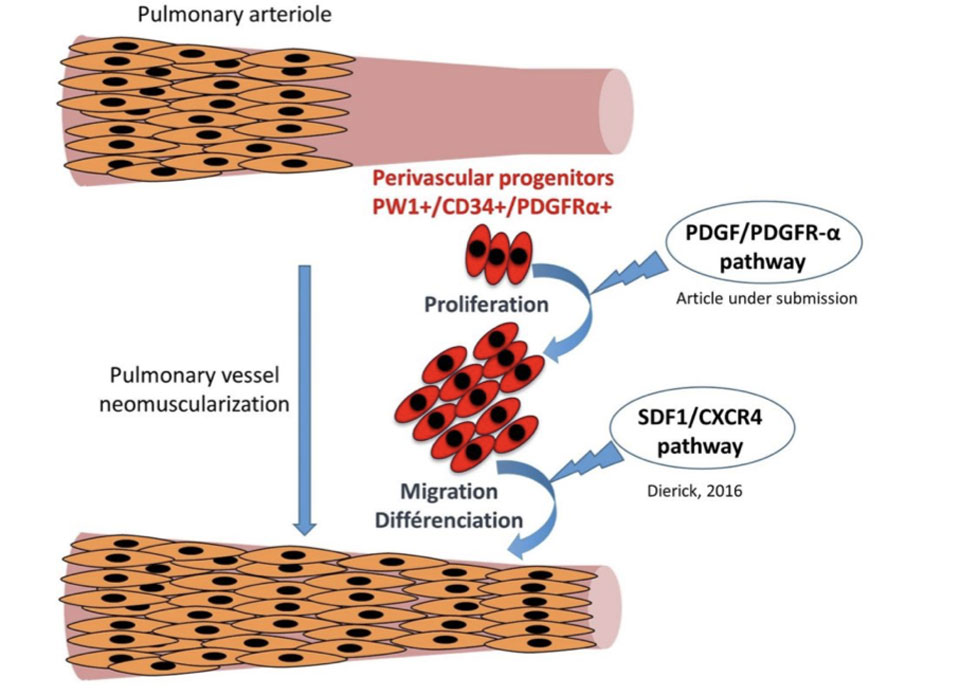
Figure 4. Pulmonary arterioles are mainly non muscularized and neomuscularization is a hallmark of pulmonary hypertension. Our data show that pulmonary vascular progenitor cells are recruited by PDGFRα signalling activation and differenciate into new smooth muscle cells following CXCR4 signalling.

Figure 5. Progenitor cells in culture differentiate into smooth muscle cells identified by SM-MHC (red) and α-SMA (green) expression.
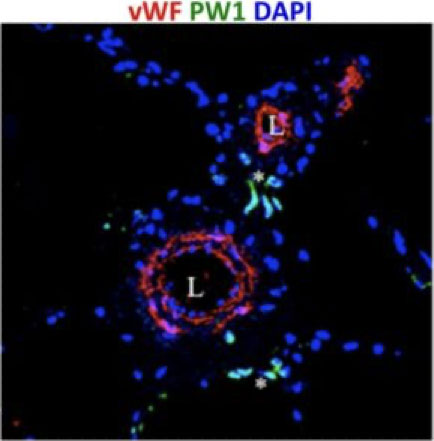
Figure 6. PW1+ Progenitor cells (green) are clustered around vessels (red) in control human lung.
2/ Our group, in tight link with the clinical genetics laboratory of the campus hospital (Pitié-Salpêtrière hospital), has identified genes involved in the genetic predisposition to pulmonary arterial hypertension (PAH) and pulmonary veno-occlusive disease (PVOD): BMP10 and KDR as new genes for heritable PAH (Eyries, Eur Resp J 2019) and EIF2AK4 (GCN2) as the gene responsible for PVOD (Eyries, Nat Genet 2014) Figure 7. The link between complete loss of GCN2, a serine-threonine kinase, and PVOD is not elucidated and is the goal of intensive research. We generated rats carrying deletions of EIF2AK4 obtained through CrispR-CAS9 gene targeting to decipher, in vivo, the modes of initiation of the disease. We demonstrated that under basal and asparagine and glutamine deprivation induced by asparaginase administration, Gcn2-/- rats display molecular and cellular signatures in the lungs that may indicate a role for Gcn2 in immune homeostasis and provide further clues to the mechanisms of hPVOD development (Bignard, Am J Physiol Lung Cell Mol Physiol, 2023)
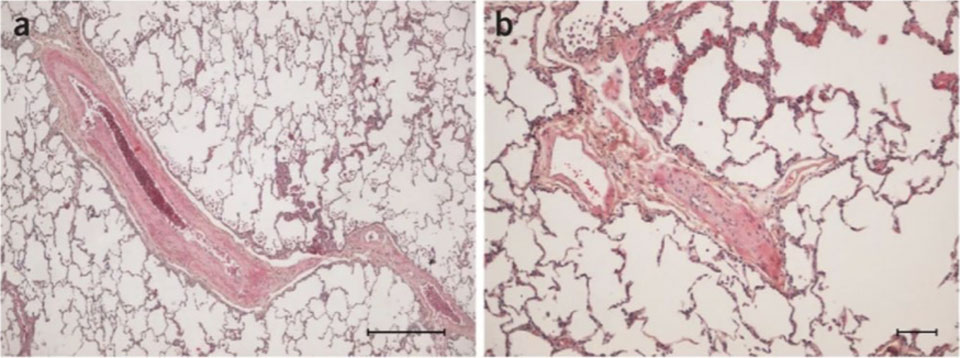
Figure 7: Pathology of heritable PVOD. (a) septal vein and (b) small vein showing intimal fibrosis and thickening of the vascular wall From Eyries and al. PMID: 24292273
Ongoing projects
Pulmonary vessels:
1/ In collaboration with the ASNR (French Nuclear Safety Authority), we are studying the effects of low doses of ionising radiation on the remodelling and function of pulmonary vessels. These low doses can affect people living in contaminated areas or nuclear workers. Epidemiological studies have shown an increase in cardiovascular disease in populations exposed to low doses of radiation, with this increase becoming more pronounced with age. We are also studying how the response varies with age. These studies are conducted on young and old mice in order to replicate human exposure.
2/ SARS-CoV-2 virus receptor ACE2 (angiotensin converting enzyme 2) is a protein expressed in vascular cells. We started a project using transgenic mice expressing human ACE2 (hACE2) to study the pulmonary vascular effects of SARS-CoV-2 infection and determine the pathways that are activated.
Systemic vessels:
3/ We are now studying the effect of obesity and metabolic syndrome on small arteries remodeling and systemic hypertension development. Our aim is to decipher the mechanisms involved in the production of new smooth muscle cells during metabolic diseases. Our hypothesis is that this remodeling is linked to perivascular progenitor cells activation. We are studying these progenitor cells and the signaling pathways that lead to their recruitment to form novel smooth muscle cells in metabolic diseases. Preventing this remodeling may help reducing the blood pressure rise that is observed under these conditions. We will study mouse models but also human samples from diabetic hypertensive and non diabetic patients.
Tools
Mouse models: We develop lineage tracing models to follow the fate of progenitor cells during pulmonary hypertension associated vascular remodeling. We use transgenic mouse models to induce or suppress candidate pathways activity in vivo following tamoxifen activation to evaluate their in the function of these progenitors and in high fat diet-induced vascular remodeling. We perform hemodynamic measurements of arterial and pulmonary arterial pressure and echo-doppler analyses of vascular and cardiac function. We use high fat diet to induce obesity, metabolic syndrome and blood pressure increase.
We analyse vascular alterations using tissue section fluorescent imaging. We study cell fate using flow cytometry and tissue imaging. We use facilities to perform single cell transcriptomic analyses and flow cytometry cell isolation and analysis. We will study patients samples using fluorescence imaging as well as spatial transcriptomics. Protein expression variations are also studied using classical western-blotting and RT-qPCR.
Collaborations
International collaborations:
Victor Chang Cardiac Research Institute, Darlinghurst, Australia, R.P. HARVEY, P. SCHOFIELD, D. CHRIST
Mc Gill University, Canada, F. DIERICK
National collaborations:
MITOVASC, Angers, D. HENRION, E. VESSIERES, L. LOUFFRANI
U999, Le Kremlin Bicêtre, C. GUIGNABERT, M. HUMBERT
Institut de Biologie Paris Seine IBPS, Paris, T. JAFFREDO
Autorité de Sûreté Nucléaire et de Radioprotection ASNR, Fontenay aux Roses, V. MONCEAU
Institut Desbrest d’Epidémiologie et de Santé Publique, Virology Department APHP, Paris, A.-G. MARCELIN, S. MAROT
Vascular Surgery department, APHP, Paris, T. COUTURE
UMS28, Paris, S. MOROSAN
UMRS 1166, Paris, F. FOUFELLE, O. BOURRON
THEME 3 – Atrial Cardiomyopathy and Atrial Fibrillation Pathophysiology – Nadine SUFFEE
Group members
Nadine SUFFEE, PhD, INSERM Researcher
Stéphane HATEM, PUPH
Eva TRENQUIER, Engineer
Raoul MANUEL, PhD, Post-Doctoral fellow
Fouad FSEIL, Master student
Ibrahim ABOU-SNEIM, Master student
Scientific context and past contributions
In the last decade clinical studies have established that adipose tissue accumulation at the atrial surface is a major risk factor for atrial fibrillation (AF), especially during metabolic disorders such as obesity (Hatem et al JACC 2016). A histomorphometry analysis has revealed 2 groups of atrial components, the one is the epicardial adipose tissue (EAT) accumulation and the other one is the fibrosis infiltration that replace this fat tissue (Figure 8A). We have identified adipokines (Activin A) secreted by the human atrial EAT that induce the fibrosis in subepicardial layer (Venteclef et al EHJ 2015). Next, we have described how EAT become fibrotic in the atrial that is a remodeling process associated with persistent AF and mediated by immune cells infiltration (Haemers et al EHJ 2017). Our investigations on the cellular origin of the adipocytes and myofibroblasts that composed the EAT and the fibrosis, respectively, revealed a common source from the progenitor cells niched in epicardial layer (EPDC, epicardial progenitor derived cells) (Figure 8B) (Suffee et al PNAS 2017). We highlighted a heterogeneous phenotype of EPDC (Figure 8C) that determined their differentiation into adipocytes or myofibroblasts in response to local factors (Suffee et al Circ Res 2020). The low concentration of atrial natriuretic peptide (ANP) secreted by atrial myocytes is a powerful adipogenic factor for the EPDCs expressing NP-receptor A through cGMP/PKG signaling pathway (Suffee et al PNAS 2017). Also, the axis AGTR1/Smad2 expressed in EPDCs induced fibrogenesis pathway in response to angiotensin-2 (Suffee et al Circ Res 2020). The accumulation of the adipose tissue is mediated through a preadipocyte factor Pref-1 expressed on EPDCs. The mechanism of Pref-1 is transient and depend on the local factors. The EPDCs-Pref1+ are reactivated with metabolic stress such as free fatty acid that engaged this precursor in the adipogenesis pathway (Azevedo et al in progress)
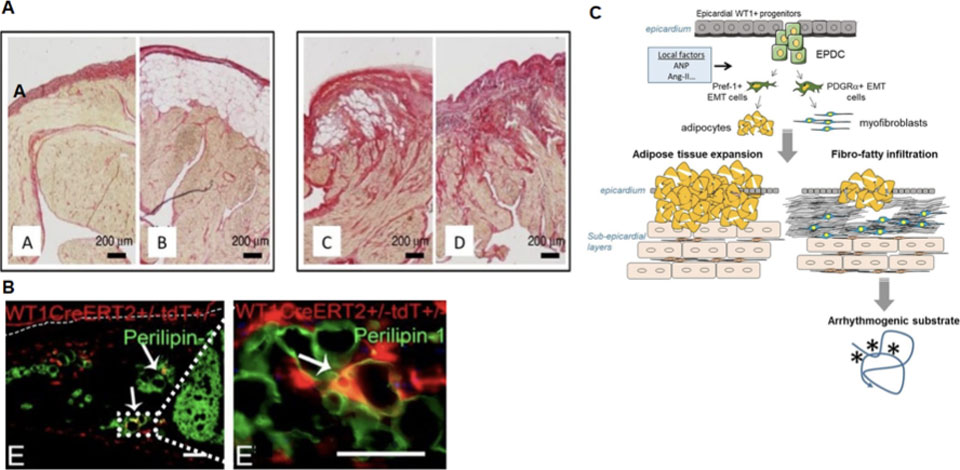
Figure 8. Epicardium is reactived early during the formation of the atrial cardiomyopathy and the substrate of atrial fibrillation. (A) The human atrial components such as epicardial adipose tissue (EAT) and fibrosis are revealed with Sirius red staining and show non-fibrotic remodelled epicardium without (A) or with (B) subepicardial adipose tissue and fibrotic remodelled epicardium with (C) or without (D) subpicardial adipose tissue. (B) The epicardial progenitor cells (EPDC) are the source of adipocytes as revealed the mouse model of lineage tracing. Coimmunostaining for perilipin-1 (adipogenic marker) and WT1-tomato (marking EPDCs) in atrial cells of mice fed a high fat diet. Arrows indicate epicardium-derived adipocytes coexpressing WT1-tomato and perilipin. (C) The recruitment of cells derived from EPDC and pre-engaged in the distinct adipocyte or fibroblastic lineages can result in the arrhythmogenic fibro-fatty infiltration of atrial subepicardial layers.
To investigate mechanisms involved in the fatty-to-fibrosis remodeling we used an obesogenic diet in mice that induced the atria cardiomyopathy (ACM) characterized with a shift of the atria metabolism in favor of long chain of fatty acid storage through β-oxidation pathway, a vulnerability to AF and an inflammatory response in atria (Suffee et al CVR 2022). This mouse model of ACM mimic fatty-to-fibrosis remodeling such observed in human tissue with immune cells infiltration (Figure 9A). The transcriptomic tools and transgenic mouse model of deletion have revealed subsets of macrophage niched in subepicardial layer (Crepin et al biorxiv 2026) (Figure 9B,C).
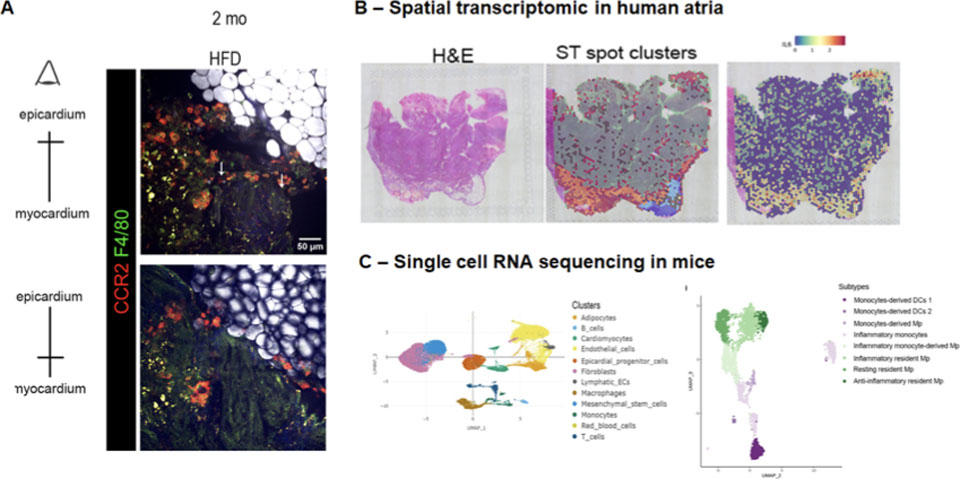
Figure 9. Mouse model of the atrial cardiomyopathy revealed immune cells infiltration in epicardium. (A) The mouse model of the atrial cardiomyopathy (ACM) induced with high fat diet mimic atria remodeling with adipose tissue accumulation, fibrosis and immune cells infiltration such as monocytes (CCR2) and macrophages (F4/80) revealed with bi-photon and Raman spectral microscopies. Tools of spatial transcriptomic used for human atria (B) and single cell RNA sequencing used for obese mice with ACM(C) revealed subsets of macrophages specifically localized in subepicardium.
In collaboration with N. Farès (Saint Joseph University Beyrouth), we have recently demonstrated that increasing myocardial atrial natriuretic peptide (ANP) availability improves heart failure in diabetic cardiomyopathy. We showed that mutated ANP (mANP) that is resistant to neprilysin degradation, reduces left ventricular dysfunction and fibrosis in the heart of type 2 diabetic rats with greater efficacy than ANP unmutated. The mANP produced higher cardiac cyclic GMP levels and stronger antifibrotic signaling in vivo than ANP. Hence, myocardial ANP bioavailability is crucial for limiting fibrosis and improving cardiac function in diabetic cardiomyopathy (Bakhos, Eur Heart J Open, 2025).
Ongoing projects
In the line to enrich the knowledge on the fatty-to-fibrosis remodeling, our projects are structured in 4 aims:
The migration of resident macrophages to the EPDCs suggests crosstalk between EPDCs and macrophages. To this end, we examine the impact of macrophages on the differentiation of EPDCs.
We also interest on the effect of macrophages polarized on the phenotype of EAT.
In the ACM mouse model, lymphocytes infiltrate in the first step of the disease, as observed in patients with primo-AF, whether they are obese or not. We will investigate their role in exacerbating remodelling, notably through the inflammatory response.
The immune response is observed in arrhythmogenic diseases in both the atria and the ventricle. We will identify the immune profile and we will compare it to establish an immune atlas.
Tools
We use human atria biopsies and blood samples to investigate phenotype and function of immune cells that we associate with imagery, flow cytometer, spatial transcriptomic and metabolomic tools.
We use transgenic mouse model one for lineage tracing for precursors cells and subsets of macrophage and the second one with a deficiency for specific subsets of macrophage. We perform clinical cardiac measurements and AF vulnerability test. To induce chronic ACM we used an obesogenic mouse model induced with high fat diet. To study acute ACM we use heart failure mouse model with reduced ejection fraction (HFrEF). To establish profile analysis of precursor and immune cells we use immunoassay, single cell RNA sequencing, western blot and flow cytometer. To test therapeutical strategies, we use neutralizing antibodies injected in mouse models of ACM.
Collaborations
International collaborations :
Indian Institutes of Technology, Delhi, India, I. GUPTA
CHUV, Département Cœur-Vaisseaux, Lausanne, Suisse, N. ROSENBLATT-VELIN
MAESTRIA H2020 European Consortium
Cardiovascular Research Center Massachusetts General Hospital, Boston, USA
Université Saint Joseph, Department of physiology, Beyrouth, Lebanon, N. FARES
Mayo Clinic College of Medicine, Rochester, USA, J. BURNET
National collaborations :
Institut Mondor de recherche Biomédicale, Créteil, A. BOISSONAS
Cardiac and Thoracic Surgery, APHP, Paris, G. LEBRETON, P. LEPRINCE, E. BERG, E. GANDJBAKHCH, T. ROLLAND.
Gustave Roussy, Villejuif, S. DURAND
Institut Pasteur, Paris, E. KARKENI
Autorité de Sûreté Nucléaire et de Radioprotection ASNR, Fontenay aux Roses, V. MONCEAU
INSERM U1046 – UMR CNRS 9214 – Université de Montpellier R. ANDRIANTSITOHAINA
CHU Cardiac and Thoracic Surgery, Lille, S. NINI
PARCC, Paris Centre de recherche cardiovasculaire, Paris, H. AIT-OUFELLA
Nutriomics, Paris, E.L. GAUTIER
THEME 4 – AI and Cardiovascular Pathophysiology – Stéphane HATEM
Group members
Stéphane HATEM, MD, PhD, PUPH
Co direction with Maharajah PONNAIAH Head of the ICAN I/O corelab
Ali RAMMAL, PhD student
Hind SAHABEDDINE, PhD student
Kimia SADRADDINI PhD student
Scientific context and past contributions
Artificial intelligence (AI) is reshaping the landscape of biomedical science and cardiovascular research, specifically. Indeed, AI offers to researchers the unique capacity to analyze complex sets of data, to identify new trajectories or clusters of data, leading to the identification of novel pathological processes, subpopulation of patients or risk factor.
1/ Machine learning approach to identify clusters of patients with distinct cardiac chronotropic response during exercise
We are developing machine learning algorithm to analyze the chronotropic cardiac response to an incremental exercise. Using AI-based unsupervised hierarchical clustering and algorithmic analysis of HR trajectory to identify subpopulations of patients with cardiopathies and heart failure. Then, using different physiological approaches we are deciphering pathophysiological mechanisms underlying patient clustering.
2/ Machine learning models of cardiac echo and MRI imaging
We have developed an algorithmic tool combining both cardiac imaging with magnetic resonance and machine learning methods to develop novel biomarkers to improve atrial fibrillation diagnosis. We measured atrioventricular groove adipose tissue characteristics during routine CMR and proved that it can be used as a proxy of atrial epicardial adipose tissue deposit. This was integrated into a multi-parametric CMR biomarker for early identification of atrial cardiomyopathy (Bialobroda, Eur Heart J Imaging Methods Pract, 2024). This is a novel development for cardiac imaging analysis using AI. We had previously used MRI to quantify different left atria myocardial components and we were able to correlate total, interstitial and fatty fibrosis the with 2D histology (Bouazizi, Plos One, 2018).
Ongoing projects
1/ We are currently studying the mechanisms leading to ANP protective effects on cardiac fibrosis using animal models of Type 2 diabetes-induced heart failure to provide novel therapeutic options in cardiometabolic diseases.
2/ Machine learning approaches are currently been developed to improve cardiac disease diagnosis and prognosis and to provide novel patient stratification tools.
Tools
– Large sets of Data (echo CPET) obtained from patient cohorts with clinical follow-up
– Computational infrastructure for data storage, mining and analysis
– AI expertise (ICAN I/O, SCAI-SU and SU Abu Dabhi)
Collaborations
International collaborations :
Mayo Clinic College of Medicine, Rochester, USA, J. BURNET
National collaborations :
Laboratoire d’Imagerie Biomédicale, U 1146, Paris, A. REDHEUIL

| Name | Position | ORCID |
|---|
- Bakhos, JJ, Saliba, Y, Hajal, J, Achkouty, G, Oskaridjian, H, Albuquerque, M et al.. Inhibiting atrial natriuretic peptide clearance reduces myocardial fibrosis and improves cardiac function in diabetic rats. Eur Heart J Open. 2025;5 (2):oeaf031. doi: 10.1093/ehjopen/oeaf031. PubMed PMID:40201591 PubMed Central PMC11977460.
- Bialobroda, J, Bouazizi, K, Ponnaiah, M, Kachenoura, N, Charpentier, E, Zarai, M et al.. The epicardial adipose tissue confined in the atrioventricular groove can be used to assess atrial adipose tissue and atrial dysfunction in cardiac magnetic resonance imaging. Eur Heart J Imaging Methods Pract. 2024;2 (1):qyae057. doi: 10.1093/ehjimp/qyae057. PubMed PMID:39224099 PubMed Central PMC11367945.
- Bignard, J, Atassi, F, Claude, O, Ghigna, MR, Mougenot, N, Abdoulkarim, BS et al.. T-cell dysregulation and inflammatory process in Gcn2 (Eif2ak4-/-)-deficient rats in basal and stress conditions. Am J Physiol Lung Cell Mol Physiol. 2023;324 (5):L609-L624. doi: 10.1152/ajplung.00460.2021. PubMed PMID:36852942 .
- Solinc, J, Raimbault-Machado, J, Dierick, F, El Bernoussi, L, Tu, L, Thuillet, R et al.. Platelet-Derived Growth Factor Receptor Type α Activation Drives Pulmonary Vascular Remodeling Via Progenitor Cell Proliferation and Induces Pulmonary Hypertension. J Am Heart Assoc. 2022;11 (7):e023021. doi: 10.1161/JAHA.121.023021. PubMed PMID:35348002 PubMed Central PMC9075467.
- Suffee, N, Baptista, E, Piquereau, J, Ponnaiah, M, Doisne, N, Ichou, F et al.. Impacts of a high-fat diet on the metabolic profile and the phenotype of atrial myocardium in mice. Cardiovasc Res. 2022;118 (15):3126-3139. doi: 10.1093/cvr/cvab367. PubMed PMID:34971360 .
- Melgari, D, Barbier, C, Dilanian, G, Rücker-Martin, C, Doisne, N, Coulombe, A et al.. Microtubule polymerization state and clathrin-dependent internalization regulate dynamics of cardiac potassium channel: Microtubule and clathrin control of KV1.5 channel. J Mol Cell Cardiol. 2020;144 :127-139. doi: 10.1016/j.yjmcc.2020.05.004. PubMed PMID:32445844 .
- Suffee, N, Moore-Morris, T, Jagla, B, Mougenot, N, Dilanian, G, Berthet, M et al.. Reactivation of the Epicardium at the Origin of Myocardial Fibro-Fatty Infiltration During the Atrial Cardiomyopathy. Circ Res. 2020;126 (10):1330-1342. doi: 10.1161/CIRCRESAHA.119.316251. PubMed PMID:32175811 .
- Eyries, M, Montani, D, Girerd, B, Favrolt, N, Riou, M, Faivre, L et al.. Familial pulmonary arterial hypertension by KDR heterozygous loss of function. Eur Respir J. 2020;55 (4):. doi: 10.1183/13993003.02165-2019. PubMed PMID:31980491 .
- Beuriot, A, Eichel, CA, Dilanian, G, Louault, F, Melgari, D, Doisne, N et al.. Distinct calcium/calmodulin-dependent serine protein kinase domains control cardiac sodium channel membrane expression and focal adhesion anchoring. Heart Rhythm. 2020;17 (5 Pt A):786-794. doi: 10.1016/j.hrthm.2019.12.019. PubMed PMID:31904424 .
- Keck, M, Flamant, M, Mougenot, N, Favier, S, Atassi, F, Barbier, C et al.. Cardiac inflammatory CD11b/c cells exert a protective role in hypertrophied cardiomyocyte by promoting TNFR2- and Orai3- dependent signaling. Sci Rep. 2019;9 (1):6047. doi: 10.1038/s41598-019-42452-y. PubMed PMID:30988334 PubMed Central PMC6465256.
- Eyries, M, Montani, D, Nadaud, S, Girerd, B, Levy, M, Bourdin, A et al.. Widening the landscape of heritable pulmonary hypertension mutations in paediatric and adult cases. Eur Respir J. 2019;53 (3):. doi: 10.1183/13993003.01371-2018. PubMed PMID:30578383 .
- Launay, T, Momken, I, Carreira, S, Mougenot, N, Zhou, XL, De Koning, L et al.. Acceleration-based training: A new mode of training in senescent rats improving performance and left ventricular and muscle functions. Exp Gerontol. 2017;95 :71-76. doi: 10.1016/j.exger.2017.05.002. PubMed PMID:28479388 .
- Suffee, N, Moore-Morris, T, Farahmand, P, Rücker-Martin, C, Dilanian, G, Fradet, M et al.. Atrial natriuretic peptide regulates adipose tissue accumulation in adult atria. Proc Natl Acad Sci U S A. 2017;114 (5):E771-E780. doi: 10.1073/pnas.1610968114. PubMed PMID:28096344 PubMed Central PMC5293064.
- Eichel, CA, Beuriot, A, Chevalier, MY, Rougier, JS, Louault, F, Dilanian, G et al.. Lateral Membrane-Specific MAGUK CASK Down-Regulates NaV1.5 Channel in Cardiac Myocytes. Circ Res. 2016;119 (4):544-56. doi: 10.1161/CIRCRESAHA.116.309254. PubMed PMID:27364017 .
- Dierick, F, Héry, T, Hoareau-Coudert, B, Mougenot, N, Monceau, V, Claude, C et al.. Resident PW1+ Progenitor Cells Participate in Vascular Remodeling During Pulmonary Arterial Hypertension. Circ Res. 2016;118 (5):822-33. doi: 10.1161/CIRCRESAHA.115.307035. PubMed PMID:26838788 .
- Haemers, P, Hamdi, H, Guedj, K, Suffee, N, Farahmand, P, Popovic, N et al.. Atrial fibrillation is associated with the fibrotic remodelling of adipose tissue in the subepicardium of human and sheep atria. Eur Heart J. 2017;38 (1):53-61. doi: 10.1093/eurheartj/ehv625. PubMed PMID:26612579 .
- Venteclef, N, Guglielmi, V, Balse, E, Gaborit, B, Cotillard, A, Atassi, F et al.. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo-fibrokines. Eur Heart J. 2015;36 (13):795-805a. doi: 10.1093/eurheartj/eht099. PubMed PMID:23525094 .
- Germain, M, Eyries, M, Montani, D, Poirier, O, Girerd, B, Dorfmüller, P et al.. Genome-wide association analysis identifies a susceptibility locus for pulmonary arterial hypertension. Nat Genet. 2013;45 (5):518-21. doi: 10.1038/ng.2581. PubMed PMID:23502781 PubMed Central PMC3983781.
Blandin C, Dilanian D, Fontaine V, Mougenot N, Gravez B, Bobin P, Duboscq-Bidot L, Farhi D, Chardonnet S, Nadaud S, Sanchez-Alonzo JL, Shevchuk A, Gorelik J, Gandjbakhch E, Hatem SN, Villard E, Balse E. Depleting trafficking regulator CASK promotes intercalated disc organization and ventricular function. BioRxiv. 2024. doi.org/10.1101/2024.10.14.618172
Prix Alain Castaigne – 2023
Stéphane Hatem
Alain Castaigne Award – 2018
Florent Soubrier
Jean Valade Award – 2014
Lamonica Cardiology Award – Académie des Sciences – 2017
France Diérick
Marion Elizabeth Brancher Award – 2017

